|
Транскрипционный анализ клеток линии HELA - продуцентов рекомбинантного пептидогликан-распознающего белка PGLYRP1 на разных стадиях развития инфекции Chlamydia trachomatis
Федеральный научно-клинический центр физико-химической медицины, Ключевые слова: Chlamydia trachomatis; PGLYRP; рекомбинантные белки; транскриптомика DOI: 10.18097/BMCRM00113 ВВЕДЕНИЕ
Пептидогликан-распознающие белки человека (PGLYRP) являются компонентами врожденного иммунитета, обладающими антибактериальной активностью [1]. PGLYRP обнаружены у всех млекопитающих и некоторых других таксономических групп [2]. У млекопитающих описано четыре белка (PGLYRP1-4), которые были идентифицированы как бактерицидные белки [3]. Показано, что PGLYRP распознают не только бактериальный пептидогликан, но и липополисахариды в наружной мембране грамотрицательных бактерий [4]. Эти белки проявляют бактерицидную активность относительно различных видов бактерий in vitro и in vivo [5]. Механизм действия пептидогликан-распознающих белков, описанный в 2010 г., включает их связывание с пептидогликаном клеточной стенки бактерий или липополисахаридами наружной мембраны и активацию защитной системы ответа на стресс у бактерий; это приводит к гибели клетки вследствие гиперактивации данной системы [6]. В экспериментах на грамположительных и грамотрицательных бактериях связывание PGLYRP с клеточной стенкой (Bacillus subtilis) или мембраной (Escherichia coli) приводило к активации двухкомпонентных систем ответа на стресс [7]. Отдельный интерес представляло изучение активности PGLYRP против таких внутриклеточных патогенных паразитов, как Chlamydia trachomatis в связи с особенностями жизненного цикла и строением клетки этой бактерии. Жизненный цикл хламидий характеризуется существованием двух форм: инфекционных, метаболически неактивных элементарных телец (ЭТ), и неинфекционных, метаболически активных ретикулярных телец (РТ) [8]. Обе формы бактерий содержат в составе мембран липополисахариды [9], недавно в клеточной стенке был обнаружен пептидогликан [10]. Ранее нами были получены рекомбинантные PGLYRP человека и продемонстрирована их антихламидийная активность [11]. Было показано, что эти рекомбинантные PGLYRP не только ингибируют развитие хламидийной инфекции при инкубации элементарных телец в растворе с рекомбинантными PGLYRP, но и уменьшают инфекционную способность дочерних элементарных телец [11]. Однако эти данные не позволяют оценить, на каком этапе проявляется антихламидийная активность PGLYRP. Целью данного исследования было изучение изменения уровня экспрессии генов в линии клеток, продуцирующих рекомбинантный PGLYRP1 в ответ на заражение их элементарными тельцами C. trachomatis. В качестве контроля использовали клеточную линию, в которой встроен дефектный ген PGLYRP1. МАТЕРИАЛЫ И МЕТОДЫ Клеточные линии и бактериальные штаммы В работе использовали штамм E.coli TOP10 («Invitrogen», США), генотип F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(StrR) endA1 λ-, элементарные тельца штамма C .trachomatis D/UW-3/Cx (ATCC VR-885), очищенные путем ультрацентрифугирования в градиенте урограффина [12]. В работе использовали клеточную линию HeLa (ATCC CCL-2, США). Культивирование эукариотических клеток Клетки линии HeLa культивировали в среде DMEM high glucose + pyruvate + Glutamax («Thermo Fisher Scientific», США) с 10% инактивированной эмбриональной телячьей сывороткой («Thermo Fisher Scientific») и гентамицином («Thermo Fisher Scientific») 10 мкг/мл, при температуре 37°С в присутствии 5% CO2. При моделировании хламидийной инфекции клетки HeLa рассевали в 24-луночные планшеты («Corning», США) в количестве ≈105 клеток на лунку и культивировали в течение 24 ч до достижения 90% конфлюентности. Клетки заражали элементарными тельцами C. trachomatis с множественностью инфекции от 0.9 до 1 ВОЕ/клетку (включение образующих единиц). Клетки центрифугировали в течение 1 ч при 900 g. После центрифугирования клетки культивировали в термостате при температуре 37°С в присутствии 5% CO2 . Полимеразная цепная реакция Полимеразную цепную реакцию (ПЦР) проводили в объеме 25 мкл с использованием 1 – 10 нг ДНК; 5 пмоль каждого праймера, Thersus полимеразы (Евроген, Россия) с соответствующим буферным раствором. Режим амплификации: 95°С/120 с – 1 цикл; 95°С/15 с, 60°С/15 с, 72°С/60 с на 1.5 т.п.н. – 25 циклов; 72°С/120 с – 1 цикл. ПЦР-фрагменты разделяли в 1% агарозном геле и выделяли из геля набором DiaGene («Диа-М», Россия). Конструирование плазмиды с дефектным геном PGLYRP1 Для получения плазмиды с дефектным PGLYRP1 был использован сайт-направленный мутагенез. Для этого использовали вектор pEGFP-N1-PGLYRP1, полученный ранее [11]. C помощью праймеров pg1f_hind /pg1stpR и pg1stpF/pg1r_sal (дополнительные материалы, табл. 1) были амплифицированны два фрагмента. Для сборки фрагмента из двух частей, 5 нг каждого из выделенных фрагментов с первого этапа смешивали, добавили смесь для ПЦР за исключением праймеров. Программа амплификации: первичная денатурация 120 с при 95°С, 10 циклов в режиме: 95°С – 10 с, 60°С – 10 с, 72°С – 60 с. Далее, были добавлены праймеры pg1f_hind/ pg1r_sal, после чего было проведено еще 20 циклов в режиме: 95°С – 10 с, 60°С – 10 с, 72°С – 60 с. В результате был получен ПЦР-фрагмент, соответствующий структурной части гена PGLYRP1 с мутацией G117A, приводящей к аминокислотной замене W39Stop. Полученный полноразмерный фрагмент выделяли из геля, расщепляли эндонуклеазами рестрикции HindIII и SalI («Thermo Fisher Scientific»). Реципиентную плазмиду pEGFP-N1-PGLYRP1 обрабатывали теми же эндонуклеазами, лигировали с полученным фрагментом лигазой фага Т4 («Thermo Fisher Scientific»,) и трансформировали лигазной смесью клетки E. coli Top10, которые высевали на чашки Петри с агаризованной средой LB, содержащей 30 мкг/мл канамицина («Sigma», США). Выросшие колонии анализировали путем ПЦР с использованием олигонуклеотидов N1 и N2 (дополнительные материалы, таблица 1) Колонии, содержащие плазмиды со вставкой, высевали в жидкую среду LB, затем из культуры выделяли плазмидную ДНК. Последовательность плазмид подтверждали методом циклического секвенирования по Сэнгеру с использованием капиллярного секвенатора AbiPrism 3730xl («Applied Biosystems»,США). Полученная плазмида получила название pEGFP-N1-PGLYRPm (m – мутантная). Получение линии клеток HeLa – продуцента PGLYRP1 Для трансфекции клеток HeLa использовали плазмиды pEGFP-N1-PGLYRP1 и pEGFP-N1-PGLYRPm, выделенные препаративно с помощью набора Qiagen Plasmid Maxi Kit («Qiagen», США). Для трансфекции клетки высевали в лунки 24-луночного планшета по 105 в лунку (так, чтобы во второй день эксперимента достичь 80-90% конфлюэнтности). Во второй день эксперимента клетки трансфицировали с помощью набора Lipofectamine 3000 («Thermo Fisher Scientific»), используя 1 мкг плазмидной ДНК на лунку 24-луночного планшета, количества используемых реактивов Lipofectamine 3000 и Р3000 составляли по 0.5 мкл на лунку. В пробирке №1 ДНК смешивали с реагентом Р3000 и доводили до объема 25 мкл средой Opti-MEM. В пробирке №2 Lipofectamine 3000 также доводили до объема 25 мкл средой Opti-MEM. Далее добавляли по каплям содержимое пробирки №1 к содержимому пробирки №2, перемешивали пипетированием несколько раз. После 5-минутной инкубации при комнатной температуре полученные комплексы в объёме 50 мкл по каплям добавляли к клеткам. Через 48 ч клеткам меняли среду на DMEM с 500 мкг/мл антибиотика G418 («Gibco», США) и культивировали в течение 2-3 недель до гибели всех нетрансфицированных клеток в контрольной лунке. Выжившие клоны переносили в новый планшет для дальнейшего культивирования и анализа. Выделение РНК Выделение суммарной РНК проводили из культуры клеток реактивом для выделения суммарной РНК ExtractRNA («Евроген»,) из расчета 1 мл реагента на 10 см2 поверхности культуральной посуды согласно инструкции производителя. Количество выделенной суммарной РНК определяли с помощью Qubit® 2.0 Fluorometer («Life Technologies», США) с использованием набора Quant-iT™ RNA Assay Kit, 5–100 ng («Thermo Fisher Scientific»). Суммарную РНК (5 мкг в образце) обрабатывали ДНКазой I (2 единицы активности (е.а.)) («Thermo Fisher Scientific») в присутствии 20 е.а. ингибитора рибонуклеаз в течение 30 мин при 37°С, далее была проведена фенол-хлороформная экстракция с последующим осаждением этанолом по стандартному протоколу [13]. Реакция обратной транскрипции Для проведения реакции обратной транскрипции использовали набор RevertAid RT Reverse Transcription Kit («ThermoFisher Scientific»), олигонуклеотиды, комплементарные поли-А последовательности (Т20), дезоксинуклеозидтрифосфаты. Для реакции обратной транскрипции использовали 1 мкг суммарной РНК, 100 пмоль праймера Т20, 20 е.а ингибитора рибонуклеаз, 200 е.а. обратной транскриптазы. Компоненты реакции смешивали согласно рекомендациям производителя и инкубировали в течение часа при 42°С, а затем инактивировали ферменты в течение 5 мин при 70°С. Полученную кДНК использовали для амплификации структурной части гена PGLYRP1 и последующего секвенирования. Вестерн-блот гибридизация Образцы культуральной жидкости линий HeLa-PGLYRP1 и HeLa-PGLYRP1m осветляли центрифугированием (300 g, 10 мин), добавляли 4-х кратный буферный раствор Лэммли, инкубировали 5 мин при 95°С. Образцы разделяли в 13,5%-SDS- полиакриламидном геле в трис-глициновом буферном растворе (25 мМ Tris-OH, 250 мМ глицин, 0,1% SDS). Далее проводили полусухой перенос белков на PVDF мембрану («Amersham Biosciences», США) при 1 мA на 1 см2 геля в течение 30 мин. Мембрану инкубировали в PBST (PBS, 0,1% Tween-20) с 5% молоком («BioRad», США) 30 мин и инкубировали в течение 1 ч при комнатной температуре с первичными антителами на полигистидиновую последовательность (Takara, США) в разведении 1:10000. После трех промывок в PBST, мембрану инкубировали в течение 1 ч при комнатной температуре со вторичными антителами овцы против иммуноглобулинов G мыши, меченными пероксидазой хрена («Amersham Biosciences») в разведении 1:10000. Мембрану проявляли c использованием набора ECL Plus («Amersham Biosciences») согласно инструкции производителя. Сигнал детектировали помощью системы гель-документирования ChemiDoc MP («BioRad»). Транскриптомное профилирование Для амплификации мРНК с помощью набора Illumina TotalPrep RNA Amplification Kit («Ambion», США) использовали 500 нг суммарной РНК согласно рекомендациям производителя. Количество полученной РНК определяли с помощью Qubit® 2.0 Fluorometer. Для оценки экспрессии по протоколу Illumina Direct Hybridization Whole-Gene Expression Assay использовали 750 нг полученной кРНК. Использовали биочипы HumanHT-12 v4 Expression BeadChip («Illumina», США), систему iScan («Illumina») и программное обеспечение Genome Studio v1.9.0 в соответствии с инструкциями производителя. Биоинформатический анализ данных Первичную обработку данных проводили с помощью программы GenomeStudio Gene Expression Module («Illumina»), используя пакет R (R version 3.1.0 (2014-04-10)) [14]. Обработку данных проводили в оболочке R-Studio Desktop 0.99.903. Схожесть между образцами определяли на основании корреляции по Спирмену. Далее проводили вычисление среднего значения сигнала для каждого гена, после чего высчитывали значение дифференциальной экспрессии каждого гена. Вычисление дифференциальной экспрессии проводили по формуле (1), где FC – изменение уровня экспрессии гена, Xo и Xc – среднее значение уровня экспрессии гена в исследуемом образце и в контроле соответственно.
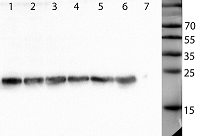
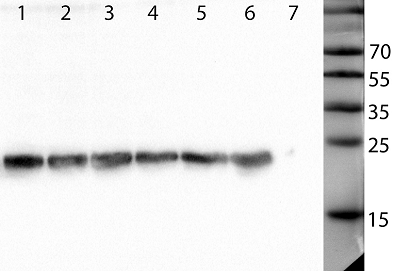
Для упрощения интерпретации данных для каждой временной точки использовали свой контроль – транскрипционный профиль незараженной линии PGLYRP1m. Поиск генов, экспрессия которых достоверно отличается в группах образцов осуществляли при помощи t-критерия Стьюдента с поправкой на множественность сравнений методом FDR по Бенжамини-Ходжбергу [15], Для визуализации использовали пакеты ggplot2 [16] и RcolorBrewer [17], а для построения диаграмм Венна был использован скрипт Venn diagram generator [18]. Функциональная аннотация дифференциально экспрессирующихся генов в терминах Gene Ontology (GO) была проведена с использованием сервиса WEB-based GEne SeT AnaLysis Toolkit. МАТЕРИАЛЫ И МЕТОДЫ Получение линий продуцентов PGLYRP1 и детекция белка Выросшие клоны после селекции на G418 анализировали путем амплификации фрагмента структурной части гена PGLYRP1. Положительные клоны анализировали на наличие мРНК путем реакции обратной транскрипции и детекции целевого белка при помощи вестерн-блот гибридизации. Из 12 отобранных клонов для каждой из линий HeLa-PGLYRP1 и HeLa-PGLYRP1m было отобрано по 3 клона, содержащих целевой участок. Для дальнейшей работы было выбрано по одному клону. Для детекции рекомбинантного белка образцы культуральной жидкости отбирали на следующий день после пересева клеток и по достижению полного покрытия лунки. В качестве контроля использовали ранее выделенный рекомбинантный PGLYRP1 с известной концентрацией. В результате рекомбинантный белок PGLYRP1 был обнаружен в культуральной жидкости линии HeLa-PGLYRP1, но не в культуральной жидкости линии HeLa-PGLYRP1m (рис. 1). Во всех трех клонах количество PGLYRP1 было одинаковым. С помощью программного обеспечения ImageJ был проведен денситометрический анализ электрофореграммы для примерного определения концентрации PGLYRP1 в культуральной жидкости. В качестве сравнения использовали рекомбинантный PGLYRP1 с известной концентрацией. Было показано, что продукция PGLYRP1 во всех клонах соответствует 0.1 мг/л культуральной среды. В результате была отобрана клеточная линия HeLa, секретирующая рекомбинантный PGLYRP1 человека, и линия, несущая встроенный дефектный ген PGLYRP1m.
Транскриптомное профилирование Для выполнения транскриптомного профилирования были использованы клеточные линии HeLa-PGLYRP1 и HeLa-PGLYRP1m. Клетки были рассеяны в 24-луночные планшеты и заражены элементарными тельцами C. trachomatis. Контролем служили клетки, которые не подвергались заражению. После заражения клеток центрифугированием клетки инкубировали при температуре 37°С в присутствии 5% СО2 в течение 1 ч, 24 ч и 72 ч, после чего проводили выделение суммарной РНК из клеток. На первом этапе подготовки образцов из клеток было выделено 8 – 15 мкг суммарной РНК. После обработки образцов ДНКазой I были получены образцы, содержащие по 2 – 4 мкг суммарной РНК без ДНК-примесей. В ходе амплификации матричной РНК с помощью набора Illumina TotalPrep RNA Amplification Kit были получены кРНК-библиотеки (РНК, комплементарная дцДНК, полученной в ходе обратной транскрипции) с выходом 1 – 15 мкг. После сканирования сигналов флуоресценции с зондов микрочипа получали первичные данные в виде таблицы с интенсивностями флуоресценции всех зондов, достоверностью детекции сигнала (p-value) по каждому из 12 образцов с одного чипа. Образцам были присвоены рабочие названия (табл. 1). Для каждого образца был сделан биологический повтор.
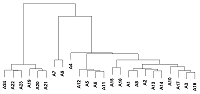
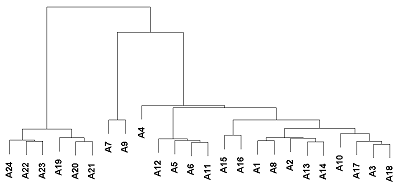
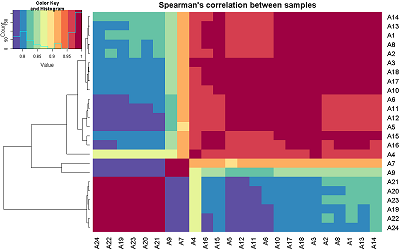
На первом этапе был проведен кластерный анализ образцов (рис. 2). Для этого из всех полученных данных сигналов флуоресценции были отсеяны те гены, уровень значимости сигнала которых был больше p>0.05. Этот уровень значимости был рассчитан в программе GenomeStudio после усреднения значения уровня сигнала для каждого гена (4 – 5 зондов на ген). Так же был проведен корреляционный анализ данных после фильтрации, результаты которого были представлены в виде тепловой карты (рис. 3).
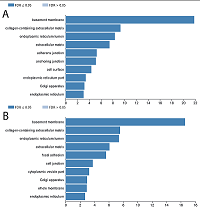
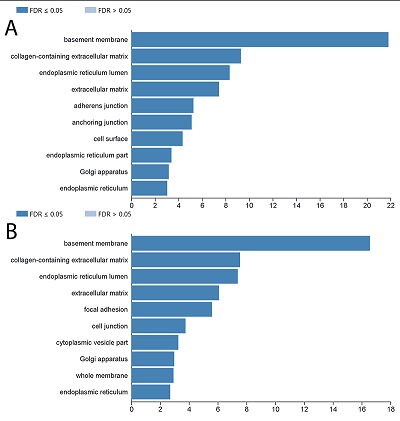
Аннотация дифференциально экспрессирующихся генов в терминах GO Для аннотации дифференциально экспрессирующихся генов в терминах GO с помощью скриптов WEBGESTALT отбирали дифференциально экспрессирующиеся гены, достоверно отличающиеся от контроля по модулю в 3 и более раз при уровне значимости p ≤ 0.01. Полученный список генов использовали для дальнейшего анализа. Для временной точки «1 ч» в список вошло 97 генов, включая 86 генов с увеличенным уровнем экспрессии. Для временной точки «24 ч» было определено 16 генов, включая 14 генов с увеличенным уровнем экспрессии. Для точки «72 ч» было определено 100 генов, включая 92 гена с увеличенным уровнем экспрессии (дополнительные материалы табл. 2S). Дифференциально экспрессирующиеся гены были найдены только для зараженных клеток линии HeLa-PGLYRP1m. В зараженных клетках линии HeLa-PGLYRP1 достоверно отличался от контроля только уровень самого PGLYRP1. Аннотированные данные представлены в трех группах генной онтологии: молекулярная функция, клеточный компонент и биологический процесс. Списки аннотаций были получены для двух временных промежутков: 1 ч и 72 ч (рис. 4, дополнительные материалы рис. 1S).
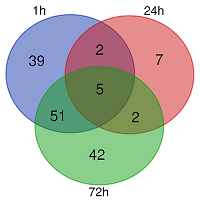
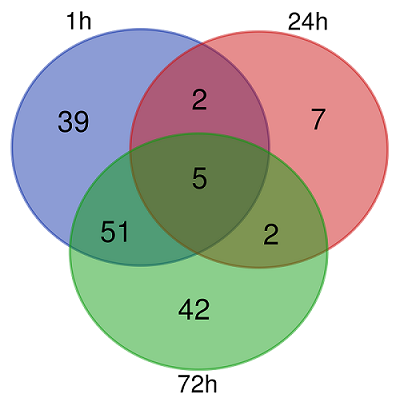
Для поиска общих дифференциально экспрессирующихся генов в клетках на разных этапах развития хламидийной инфекции были построены диаграммы Венна (рис. 5). В результате был обнаружен 51 общий ген между временными точками «1 ч» и «72 ч». При этом временной интервал «24 ч» имеет по 2 общих гена временными точками «1 ч» и «72 ч» (табл. 2).
Таким образом, нами был получен транскриптомный профиль клеток HeLa-PGLYRP1m на различных стадиях заражения C. trachomatis. ОБСУЖДЕНИЕ В данном исследовании была получена стабильная линия трансформированных клеток HeLa, продуцирующая и секретирующая рекомбинантный PGLYRP1 с выходом ≈0.1 мг/л культуральной среды. Ранее нами были получены рекомбинантные белки PGLYRP в клетках линии Expi293, синтезируемые в виде слитых белков с легкой цепью IgG с выходом 50 мг/л культуральной среды и была определена минимальная ингибирующая концентрация (МИК) при которой ингибируется развитие >90% хламидийных включений [11]. По данным вестерн-блот гибридизации и денситометрического анализа была определена концентрация целевого белка в культуральной среде клеточных линий HeLa-PGLYRP1 (рис. 1). При заражении клеток, покрывающих 90% поверхности лунки в 0,5 мл культуральной среде было 50 нг PGLYRP1, что практически соответствует МИК (200 нг/мл). Точный механизм действия PGLYRP на C. trachomatis неизвестен. Ранее нами было показано, что PGLYRP1 связывается с ЭТ хламидий. Известно, что мембрана ЭТ непроницаема для большинства молекул (крупные белки) [19]. Поскольку ЭТ являются метаболически неактивными, двухкомпонентная система ответа на стресс не включается. Было предположено, что ЭТ вместе с PGLYRP1 проникают внутрь клетки и уже там проявляется антибактериальная активность целевого белка. Для подтверждения этого было предложено изучить изменение экспрессии генов в линиях, продуцирующих рекомбинантный PGLYRP1 в ответ на заражение их элементарными тельцами C. trachomatis с помощью транскриптомного анализа. На первом этапе транскриптомного профилирования после извлечения первичных данных был проведен кластерный анализ. В результате было выделено две группы, причем в одну группу попали образцы А19-А24, что соотвествует данным для клеточной линии HeLa-PGLYRP1m, зараженных C.trachomatis. Во вторую группу попали образцы A1-A18, что соответствует данным для клеточной линии HeLa-PGLYRP1, зараженной C.trachomatis и незараженных линий HeLa-PGLYRP1 и HeLa-PGLYRP1m. Последующий корреляционный анализ подтвердил разделение образцов на две группы, с коэффициентом корреляции близкому к единице (0.95-1) внутри группы и коэффициентом корреляции 0,75-0,8 между группами (рис. 4). Как на рисунке 1, так и на рисунке 2 можно выделить группу А7, А9 с коэффициентом корреляции 0.85-0.9, однако никаких достоверных отличий от основной группы не было обнаружено. Проведенный поиск дифференциально экспрессирующихся генов и последующий анализ показали, что дифференциально экспрессирующиеся гены присутствуют только в клеточной линии HeLa-PGLYRP1m, зараженной C. trachomatis (при установленном трехкратном значении изменения уровня экспрессии по модулю). Таким образом, было подтверждено, что транскрипционный профиль зараженных клеток HeLa-PGLYRP1m отличается от остальных образцов на всех трех временных промежутках. На основании выбранных генов была построена диаграмма. Исходя из полученных данных видно, что в группах «1 ч» и «72 ч» после заражения обнаружен 51 общий ген. В жизненном цикле хламидий серовара D выход дочерних элементарных телец приходится как раз на 72 ч [20]. Таким образом, наблюдается схожий транскриптомный профиль у клеток, подвергающихся первичному заражению (1 ч) и вторичному (72 ч). При аннотации дифференциально экспрессированных генов в терминах GO было недостаточно данных, для временного интервала 24 ч, поэтому исследовали только 1 ч и 72 ч после заражения. Известно, что механизм проникновения элементарных телец в эукариотическую клетку тесно связан с цитоскелетом клетки хозяина. На стадиях проникновения хламидии активно взаимодействуют с актином, микротрубочками и другими компонентами цитоскелкта [21]. После проникновения в клетку, включение начинает перемещаться в перинуклеарную область клетки и остается тесно связанным с аппаратом Гольджи и ЭПР [22]. При анализе дифференциально экспрессирующихся генов в терминах генной онтологии можно выделить 20, 18 и 12 генов, относящихся к ЭПР, аппарату Гольджи и цитоскелету соответственно (рис. 4). Из-за прочной взаимосвязи жизненного цикла хламидии с цитоскелетом клетки-хозяина, изменение уровня экспрессии генов, вовлеченных в организацию цитоскелета, может сказываться на развитии хламидийной инфекции. Ранее нами была продемонстрирована антихламидийная активность рекомбинантных PGLYRP1 человека, в результате чего существенно сокращалось количество хламидийных включений. Для уточнения механизма действия PGLYRP1 на развитие хламидийной инфекции в клетке был проведен транскриптомный анализ клеточной линии HeLa-PGLYRP1, зараженной C. trachomatis. В ходе исследования было показано, что рекомбинантный PGLYRP1 ингибирует развитие хламидийной инфекции в клетках человека, что проявляется сходным транскрипционным профилем с незараженными клетками. Так же было показано, что в контрольных клетках уже через час после заражения изменяется транскрипционный профиль. Таким образом, можно предположить, что PGLYRP1 действует на хламидийную клетку не внутри эукариотической клетки, а снаружи или во время прикрепления к мембране или в культуральной среде. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi. org/10.18097/BMCRM00113) на сайте журнала. ЛИТЕРАТУРА
|