|
Потенциальные ингибиторы протеазы 3СLpro вируса COVID-19: репозиционирование лекарств
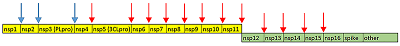
1Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича, Ключевые слова: COVID-19; протеаза; ингибитор; репозиционирование лекарств; докинг; молекулярная динамика DOI: 10.18097/BMCRM00124 ВВЕДЕНИЕ Новый коронавирус 2019 года (COVID-19) вызвал вспышку пневмонии в Ухани (Китай) в конце декабря 2019 года и быстро распространился по всему миру. Всемирная организация здравоохранения (ВОЗ) объявила эту новую вспышку коронавируса чрезвычайной ситуацией в области здравоохранения Коронавирусы, члены семейства Coronaviridae и подсемейства Coronavirinae, включают вирусы с положительной цепью РНК, которые имеют «шипы» гликопротеинов, выступающих из своих вирусных оболочек, и, соответственно, имеют форму короны [1]. Коронавирусы вызывают широкий спектр респираторных и желудочно-кишечных заболеваний у диких и домашних животных. До вспышки заболевания, вызванного вирусом COVID-19, было известно шесть штаммов коронавирусов, способных инфицировать человека, включая четыре штамма, циркулирующих ежегодно и вызывающих ОРВИ, а также два штамма, которые являются причиной острого респираторного синдрома (SARS) и ближневосточного респираторного синдрома (MERS-CoV) [1, 2]. Геном вируса COVID-19 состоит из ~30000 нуклеотидов, которые кодируют два перекрывающихся полипротеина, pp1a и pp1ab, необходимых для репликации вируса и транскрипции [3, 4]. Функциональные белки высвобождаются из полипротеинов в результате протеолиза, который главным образом, осуществляет протеазай 33,8 кДа (3СLpro) (рис. 1) [5]. Функциональное значение 3СLpro в жизненном цикле вируса вместе с отсутствием близких гомологов у людей делает этот фермент привлекательной мишенью для разработки противовирусных препаратов [6].
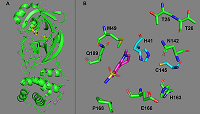
В настоящее время нет противовирусных препаратов для этой эпидемии. Недавно было сообщение, что комбинация противомалярийного препарата Гидроксихлорохина и полусинтетического антибиотика Азитромицина может оказывать терапевтический эффект, но механизм этого действия остается неизвестным [7]. Разработка новых лекарств – это длительный, дорогостоящий и сложный процесс. Одним из подходов, который может его ускорить, является поиск лекарств, которые уже были ранее одобрены для лечения других заболеваний (репозиционирование лекарств). Репозиционирование лекарств стало успешной стратегией вследствие того, что не требуются затраты времени и средств на часть обязательных этапов исследований соединений на их безопасность [8–10]. К настоящему времени уже известно несколько успешных примеров этой стратегии [11–14], и уже был проведен ряд работ по виртуальному скринингу лекарственных препаратов для поиска ингибиторов протеазы 3СLpro COVID-19 [15-18]. В данной работе был проведен виртуальный скрининг известных лекарственных средств на возможность их взаимодействия с протеазой 3СLpro вируса COVID-19. МАТЕРИАЛЫ И МЕТОДЫ Пространственные структуры протеазы 3СLpro COVID-19 в комплексах с ингибиторами были получены из банка белковых данных PDB. В качестве мишени для докинга была использована структура 5R81 (рис. 2). Место связывания потенциальных лигандов было определено на основе ковалентно-связанного ингибитора из структуры 6LU7. В качестве лигандов для докинга использовали созданную нами ранее базу данных с разрешенными к применению лекарственными препаратами, содержащими около 5 тысяч соединений. Структуры белков и лигандов были оптимизированы путем минимизации энергии с использованием метода Пауэлла с силовыми полями Tripos в вакууме с использованием пакета молекулярного моделирования SYBYL 8.1 (Tripos Inc., США). Парциальные заряды атомов белков и лигандов были рассчитаны методом Гастейгера-Хюккеля. Молекулярный докинг проводили с использованием программы DOCK 6.5 [19]. Доступная для растворителя поверхность белка была построена на основе алгоритма Коннолли с радиусом зонда, равным 1,4 Å. Электростатические и Ван-дер-Ваальсовы поля, генерируемые вокруг белка-мишени, рассчитывали с использованием сетки с расстоянием 0.3 Å. Отсечка расстояния для несвязанных взаимодействий была установлено равной 12 Å, а параметры для Ван-дер-Ваальсовых взаимодействий были взяты из набора dw_AMBER_parm99.defn. Все соединения были докированы с использованием опции оценки энергии связывания после минимизации после первоначального размещения лиганда в сайте связывания. Лучшие позы докинга были выбраны на основе оценочной функции DOCK 6.5.
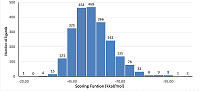
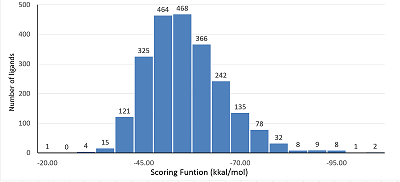
Моделирование молекулярной динамики (МД) было проведено с помощью программы AMBER 16.0. Моделирование проводилось в периодических граничных условиях с растворителем, заданным в явном виде (модель TIP3P). Для белка было использовано поле сил ff99-SB, для лигандов - parmbsc0. Перед молекулярной динамикой системы были оптимизированы методом минимизации энергии (метод крутого спуска, 1000 шагов), за которым шел постепенный нагрев до 300 К. Давление поддерживалось 1 атм. Длина продуктивной динамики была 5 нс, с шагом 2 фс. Расчет дальнодействующих электростатических взаимодействий проводили методом PME с «отсечкой» на уровне 8 Å. Температуру поддерживали с использованием динамики Ланджевена, давление — баростата Берендсена. Процедуру SHAKE использовали для поддержания постоянных длин связей с атомами водорода, с шагом интеграции 2 фс. Анализ траектории молекулярной динамики анализировали в программе VMD [20]. Расчет энергии связывания в системе белок-лиганд был сделан методом MM–GBSA (Molecular Mechanics-General Born/Surface Area). Энергия связывания MM–GBSA состоит из двух частей: энергия взаимодействия в системе белок-лиганд в газовой фазе и свободной энергии сольватации. Первая часть – сумма энергий электростатического взаимодействия и энергии Ван-дер-Вальса. Сольватационная свободная энергия включает полярную и неполярную часть. При расчете диэлектрическая константа для водной фазы была равна 80, для системы белок-лиганд была принята равной 1. Вклад неполярной части был определен с использованием расчета поверхности доступной для растворителя (solvent-accessible-surface area, SASA). РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Активный центр протеазы вируса COVID-19 представляет достаточно гидрофильную впадину, расположенную на N-концевом субдомене, в котором расположена каталитическая диада Cys145 и His41 (рис. 2). За место связывания лигандов была выбрана область вокруг ковалентно связанного ингибитора протеазы N3 из структуры pdb 5R81 . Соединения для докинга были взяты из ранее составленной нами базы данных лекарств, одобренных для клинического применения. Из этой базы были выбраны соединения с молекулярной массой в диапазоне от 250 до 1000 Да. В результате в докинге было использовано около трех тысяч соединений. Поскольку активный центр протеазы представляет собой открытую полость для большинства докированных соединений, были найдены возможные положения лигандов в нем. Селекция соединений была проведена на основе величины оценочной функции Dock (SFD) по стерической, гидрофобной и электростатической комплементарности. На рисунке 3 представлено распределение количества соединений, имеющих определенную величину оценочной функции. Видно, что максимальное количество соединений имело SFD в диапазоне 45-55 ккал/моль. Поэтому в дальнейшем рассматривали только соединения, имеющие SFD ниже -60 ккал/моль. В результате этой селекции для дальнейшего анализа были выбраны 38 соединений.
Для оценки устойчивости полученных комплексов протеаза-лиганд было проведено моделирование молекулярной динамики на траектории 5 нс, на основании которой была рассчитана энергия связывания методом MM-GBSA. Для ряда комплексов было отмечено значительное изменение положения и конформации лигандов, вплоть до выхода лиганда из активного центра, и такие комплексы были исключены из дальнейшего анализа. Для оставшихся комплексов было дополнительно рассчитано количество водородных связей и проведен анализ взаимодействия лигандов с протеазой в оптимизированных молекулярной динамики комплексах. В результате было отобрано 10 лекарств, которые потенциально могут взаимодействовать так же и с протеазой вируса. Количественные параметры для этих соединений приведены в таблице 1.
Анализ фармакологических групп и молекулярных механизмов основного действия выбранных лекарств (табл. 2) показал, что половина из них была разработана для лечения онкологических заболеваний; одно лекарство – сосудорасширяющее средство, еще одно оказывает стимулирующее действие на ЦНС. Три соединения являются ингибиторами протеаз.
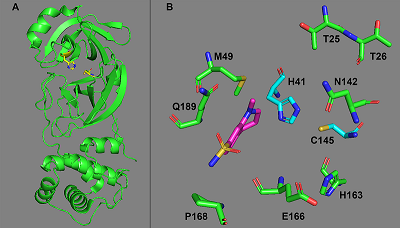
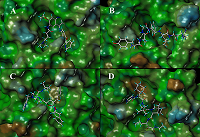
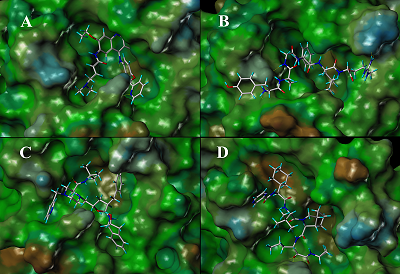
На наш взгляд, наиболее перспективными являются первые четыре препарата (рис. 4).
(1) Indinavir является ингибитором протеазы ВИЧ [21]. Ранее проведенные с помощью компьютерных методов исследования по репозиционированию лекарств для торможения протеазы 3СLpro COVID-19 выявили ингибиторы протеазы ВИЧ, в частности Saquinavir, Lopinavir и др., в том числе и Indinavir [17, 18]. (2) Telaprevir является ингибитором протеазы гепатита С. Эта фермент относится к группе сериновых протеаз [22], и разработанные ингибиторы обычно связываются с каталитическим остатком серином ковалентно; гидролиз этой ацильной связи заторможен в результате плохой доступности ёё для воды. Протеаза 3СLpro COVID-19 относится к цистеиновым протеазам со сходным с сериновыми протеазами механизмом катализа. Анализ положения Telaprevir на траектории молекулярной динамики комплекса протеазы 3СLpro COVID-19 с Telaprevir показал, что расстояние между каталитическим цистеином и карбонильной группой, которая должна участвовать в ковалентном связывании находилось в диапазоне 3-5 Å на протяжении всей траектории, что предполагает возможность образование ковалентной связи между протеазой и лекарством. (3) Противоязвенный препарат Даларгин (Dalargin) является агонистом дельта-опиоидных рецепторов и снижает синтез протеолитических ферментов. Он представляет собой гексапептид Tyr-dAla-Gly-Phe-Leu-Arg, в котором остаток аланина присутствует в виде D-изомера. В процессе подготовки данной статьи появилось сообщение, что в Научном центре биомедицинских технологий ФМБА России начаты клинические испытания этого препарата для лечения тяжелых коронавирусных пневмоний [23]. Можно предположить, что использование этого препарата будет способствовать лечению не только последствий развития вируса, но и непосредственно блокировать его размножение. (4) Еще одним перспективным препаратом является Neratinib. Он является ингибитором двойного действия, действуя в качестве ингибитора тирозинкиназы рецепторов ErB, к которым относятся рецепторы эпидермального фактора роста (epidermal growth factor receptor, EGFR), и рецептора эпидермального фактора роста 2 человека (human epidermal growth factor receptor 2, Her2). Neratinib блокирует работу рецепторных тирозинкиназ через образование ковалентной связи с остатком цистеина в активных центрах киназных доменов этих рецепторов [23]. Как указывалось выше, протеаза 3СLpro 2019-nCoV относится к цистеиновым протеазам, в активном центре которой присутствует активированный остаток цистеина. Анализ молекулярной динамики комплекса этой протеазы с Neratinib показал, что во второй половине молекулярной динамики расстояние между атомом серы и местом ковалентной пришивки стабилизируется в районе 4-5 Å, что позволяет допустить возможность образования ковалентной связи между протеазой и ингибитором, которая должна увеличить прочность комплекса. Таким образом, проведенный поиск возможных лигандов из числа разрешенных к применению лекарств позволил предложить для экспериментальной проверки лекарственные препараты, которые могут связываться с одной из основных мишеней - протеазой 3СLpro COVID-19. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов ЛИТЕРАТУРА
|