|
Обработка длинных чтений транскриптомного секвенирования на облачной вычислительной платформе Amazon Web Services 1Центр стратегического планирования и управления медико-биологическими рисками здоровью, 119121, Москва, ул. Погодинская, 10; *e-mail: g-s2011@mail.ru Ключевые слова:постгеномные технологии; транскриптом; РНК; секвенирование; биоинформатика; облачные вычисления; нанопоровое секвенирование DOI: 10.18097/BMCRM00131 ВВЕДЕНИЕ
Развитие технологий Next generarion sequencing (NGS) требует овладения базовыми биоинформатическими методами широким кругом молекулярных биологов, а не только специалистами в анализе данных. Большинство решений в области программного обеспечения (ПО) для анализа данных NGS представляют собой свободно распространяемые программы с открытым исходным кодом. Открытость кода обеспечивает его гибкость и быструю эволюцию, однако самостоятельная работа с открытым кодом часто требует нетривиальных навыков. В то же время, сервер с предустановленным программным обеспечением для обработки результатов секвенирования, может широко использоваться с применением облачных технологий [1]. В отличие от геномики, в области протеомики и метаболомики экспериментатор всегда работает на приборах с предустановленным коммерческим программным обеспечением, предназначенным для анализа, визуализации и статической обработки результатов. Однако существуют свободно-распространяемые программные продукты с графическим интерфейсом, созданные для обработки и анализа данных протеомных экспериментов: SearchGUI, MaxQuant (идентификация и полуколичественный анализ белков [2]), ХСМС-online (анализ метаболитов [3]) и др. Такого рода ПО, имеющее графический интерфейс, работающее под операционными системами (ОС) семейства Windows и предназначенное для анализа геномно-транскриптомных данных, не получило широкого распространения. В области анализа данных NGS в подавляющем большинстве случаев используют биоинформатические программы, разработанные для запуска через командную строку ОС Linux. Как правило, эти программы отработаны и протестированы для выполнения под операционной системой Ubuntu: RSEM [4], bowtie2, STAR, kallisto, сборщик геномов и метагеномов SPAdes [5-6]. Программы для обработки данных NGS, имеющие графический интерфейс UGENE («Унипро», Россия) и CLC Genomics Workbench («Quagen», США), ориентированы на нужды обучения или коммерческой биотехнологии. При переходе к работе с большими объемам данных требуется оплата лицензирования и/или платная настройка на стороне конечного пользователя. К настоящему времени использование коммерчески доступных облачных сервисов позволило «демократизировать» возможности обработки научных данных для пользователей, которые не являются специалистами в области биоинформатики. В частности, смещаются акценты приложения профессиональных навыков: при наличии единожды отлаженной последовательности действий (конвейера, протокола) рутинная обработка геномных и транскриптомных данных переходит в распоряжение экспериментатора. В данной статье предложена подробная инструкция применения технологий облачных вычислений, не требующая организации собственных дорогостоящих вычислительных решений для анализа геномов на транскриптомном уровне с применением нанопорового секвенатора и доступная широкому кругу пользователей, включая школьников и студентов. Высокопроизводительное секвенирование с использованием нанопоры Массивное параллельное секвенирование нуклеиновых кислот представлено двумя технологическими подходами в зависимости от длины фрагментов прочтений. Один из них основан на секвенировании относительно небольших (от 25 до 500 нуклеотидов) фрагментов нуклеиновых кислот с перекрывающимися последовательностями, второй – на секвенировании протяжённых фрагментов длиной несколько тысяч нуклеотидов. Ко второму подходу относится технология нанопорового секвенирования, разработанная компанией «Oxford Nanopore Technology» («ONT», Великобритания), которая в 2015 г. вывела на рынок коммерческий нанопоровый секвенатор MinION. MinION остаётся наиболее распространённым нанопоровым секвенатором, хотя компания «ONT» предлагает секвенаторы и других типов – Flongle, GridION и PromethION (https://nanoporetech.com/products). Нанопоровое секвенирование нуклеиновых кислот основано на пропускании молекул однонитевой ДНК или РНК через белковые поры диаметром 1-2 нм, формируемые рекомбинантными вариантами поринов, встроенных в искусственные амфифильные мембраны. Нуклеотиды блокируют прохождение электрического тока через пору, генерируя последовательность изменений силы тока во времени. Работа секвенатора MinION и процесс сбора первичных данных контролируется программой MinKNOW, которая доступна для авторизованных пользователей на сайте «ONT» (https://nanoporetech.com/) и может быть скачана и инсталлирована на управляющий компьютер. Программа MinKNOW позволяет наблюдать процесс секвенирования в реальном времени: мониторинг температуры камеры проточной ячейки, величину прикладываемого к нанопоре напряжения, изменение во времени количества секвенированных последовательностей, их длины, долю работающих пор и другие характеристики процесса. Программа MinKNOW регистрирует и сохраняет в формате fast5 данные о величине ионного тока через нанопоры (первичные данные), которые необходимо перевести в последовательность нуклеотидов в ходе процедуры «бейс-коллинг» или «бейз-коллинг» (от англ. base-calling). Программа MinKNOW работает на компьютерах под управлением операционной системы Windows, поэтому дальнейшее выполнение протокола ориентировано, в первую очередь, на пользователей этой операционной системы. Средняя длина секвенирования нуклеотидных последовательностей (средняя длина прочтения) в случае нанопорового секвенирования такова, что позволяет «прочитать» большинство транскриптов полностью как единичную молекулу. В рамках данной работы мы описываем пошаговый протокол обработки первичных данных, получаемых на примере анализа ткани печени человека. Исходные данные Секвенирование транскриптома на нанопоровом секвенаторе MinION требует наличия исходного материала – матричной РНК (мРНК) для приготовления используемой для секвенирования библиотеки. Доля мРНК в суммарной РНК, присутствующей в клетке, незначительна и составляет всего 2-5%. Для сохранения молекул мРНК к биообразцу следует добавлять стабилизирующий раствор RNAlater («Thermo Fisher Scientific», США), затем биообразец необходимо хранить и транспортировать при -20ºС, чтобы выход суммарной РНК составлял примерно 50-70 мкг на 100 мг ткани. Предлагаемый протокол был апробирован на данных, полученных при так называемом прямом секвенировании РНК, когда секвенируемая библиотека представляет собой гетеродуплексы матричной мРНК с комплиментарной кДНК (для получения гетеродуплексов необходимо выделить из биопробы не менее 500 нг мРНК). Приготовление библиотеки предусматривает присоединение к концам гетеродуплексов специальных моторных белков, связывающихся с белковой порой и обладающих гиразной активностью. Моторные белки при участии молекул ATP расплетают дуплексы, направляя цепь РНК в пору с контролируемой скоростью. Для приготовления библиотеки используют набор Direct RNA Sequencing Kit (SQK-RNA002, «ONT»). Процедуры проводят в соответствии с рекомендациями производителя [7]: синтез комплиментарной кДНК с помощью обратной транскриптазы, затем - лигирование адаптеров. Для очистки целевых нуклеиновых кислот от присутствующих в реакционной смеси ненужных для секвенирования компонентов (в том числе, ферментов) применяют магнитные шарики-«битсы». Первичные данные о транскриптоме клеток генерируют нанопоровым секвенатором MinION с проточной ячейкой FLO-MIN106, сохраняют в одном из форматов, предоставляемых «ONT» и депонируют в локальной системе хранения данных. В случае, если на основе данных планируется научная публикация, то файлы fast5 следует загрузить в международный ресурс Sequence Read Archive. Разработка биоинформатического протокола Мы применили подход, упомянутый в источнике [8] с целью отобрать в автоматическом режиме статьи по теме обработки транскриптомных данных методами биоинформатики. Поскольку простота и востребованность описанных в научных публикациях технических решений косвенно подтверждается частотой цитирования, мы сформировали выборку релевантных публикаций, описывающих наиболее простые в эксплуатации подходы. Были отобраны алгоритмы для первичной расшифровки сигналов секвенатора, а также алгоритмы контроля качества процедуры «бейз-коллинг» и картирования результатов расшифровки на геном человека с последующим количественным анализом уровня транскрипции секвенированных генов. «Бейз-коллинг» Наиболее часто используемым и эффективным программным пакетом для процедуры перевода сигналов нанопорового секвенатора в нуклеотидные последовательности является guppy_basecaller. Часть конкурирующих програмных продуктов либо не поддерживается производителем («Albacore Oxford Nanopore Basecaller», Великобритания), либо обладает более низкими эксплуатационными характеристиками [9]. Точность секвенирования при использовании расширенных нейросетевых моделей в guppy_basecaller достигает 99.73%. Подробно варианты использования этой программы рассмотрены в [10]. Необходимо отметить, что программное обеспечение guppy_basecaller не является свободно-распространяемым и требует от пользователя прохождения процедуры авторизации на сервере community.nanoporetech.com. Несмотря на то, что программное обеспечение для «бейз-коллинга» предустановлено на виртуальную машину, описываемую в нашей работе, его использование, в соответствии с лицензионным соглашением, требует разрешения от производителя нанопоровых секвенаторов для каждого конкретного пользователя. Целесообразность предустановки программы guppy_basecaller на виртуальную машину AWS с многопользовательским доступом обусловлена тем, что подавляющее большинство доступных пользователям локальных компьютеров, с которыми сопрягается MinION, не имеют рекомендуемого для выполнения процедуры «бейз-коллинг» графического ускорителя Nvidia V100. Контроль качества процедуры «бейз-коллинга» Для верификации результатов работы программы guppy_basecaller рекомендуется использовать программу оценки качества с графическим интерфейсом, написанную на языке программирования R – minIONQC.R [11] и распространяемую без ограничений в виде исходного программного кода. Выравнивание FASTQ файлов на геном Minimap2 - универсальная программа выравнивания для сопоставления последовательностей ДНК или мРНК с референсной базой данных. Оценка статьи [12], описывающей применения этой программы для выравнивания результатов нанопорового секвенирования на референсный геном, показала, что она была процитирована более 650 раз. Программа работает с короткими считываниями длиной ≥100 п.н., а также с геномными считываниями более 1 килобаз с частотой ошибок от 1.5%. Minimap2 позволяет проводить сборку (ассемблирование) полноразмерных считываний РНК или комплиментарной ДНК, «картировать» прочтения на последовательности протяженных участков генома (контиги) или на близкородственные хромосомы длиной в сотни мегабаз. Исходя из этого, можно утверждать, что в состав рассматриваемого в статье алгоритма, наряду с анализом транскриптома человека, заложены принципы полногеномного анализа. Намного большее количество цитирований (более 23 тысяч) имеет статья того же автора, опубликованная годом ранее [13]. Изложенная в статье работа связана с выполнением крупного геномного проекта 100-k Genomes England (www.genomicsengland.co.uk). В статье рассматривается формат данных SAM (Sequence Alignment/Map) и позволяющий его обрабатывать программный инструмент SAMtools. SAMtools влючает в себя различные утилиты, такие как индексирование, анализ мутаций и средство просмотра выравнивания, что делает его незаменимым в оценке выравнивания и пост-обработки в формате SAM. В нашей работе утилита stats из пакета SAMtools применена для контроля качества процедуры выравнивания данных нанопорового секвенирования на референсный геном или транскриптом. Количественный анализ Для определения уровня экспрессии транскриптов наиболее часто используют программу Salmon. Программа поддерживает алгоритм со сверхбыстрой процедурой считывания, что позволяет работать с характерными для нанопоры длинными прочтениями. Это повышает точность оценок при анализе дифференциальной экспрессии генов [14]. Программа Salmon работает с выравниванием как длинных, так и коротких прочтений, поэтому подходит для интерпретации данных и ONT и Illumina. В исследовании под руководством Soneson [15] применили нанопоровую ONT-технологию к секвенированию длинной нативной РНК в образцах двух линий клеток человека (HAP1 и HEK293) с целью оценки возможностей нанопоровой технологии идентифицировать и количественно определить уровень экспрессии генов. Данные прямого (без ПЦР) секвенирования кДНК по ONT-протоколу хорошо соотносились с результатами стандартного секвенирования на платформе RNA-Illumina. Кроме того, авторы показали, что программа Salmon может применяться для поиска и количественного анализа продуктов альтернативного сплайсинга. Результаты масштабного применения нанопорового секвенатора для исследования транскриптома человека опубликованы коллективом Института Джона Хопкинса, США [16] в кросс-лабораторном формате (с участием лабораторий Канады и Великобритании). В исследовании было считано 9.9 миллиона последовательностей для линии клеток человека GM12878 (линия из В-лимфоцитов поддерживается в «Corriel Institute», США) с использованием тридцати проточных ячеек MinION в шести учреждениях. Средняя длина прочтений составила 771 основание, а максимальная выровненная длина - более 21 тыс. оснований. Обработку данных этого проекта производили программой Salmon. Облачная платформа Amazon Разработчики вычислительных платформ, связанных с обеспечением бизнес-процессов на основе больших данных, используют два пути. При первом пользователь может самостоятельно собрать вычислительный кластер, для которого нужно выделить или оборудовать особое помещение, обеспечить температурный режим, установить контроль влажности и электромагнитного излучения, использовать бесперебойное снабжение серверов и коммуникационных устройств электропитанием, а также нанять квалифицированный обслуживающий персонал и нести все затраты по дальнейшему содержанию. Второй путь, рассматриваемый в данной статье, - арендовать нужные вычислительные мощности дистанционно в суперкомпьютерном центре, пользоваться только необходимым функционалом, и не нести перечисленные выше эксплуатационные расходы. Успех компаний Amazon или Alibaba Group доказывает, что у локального вычислительного кластера появилась достойная альтернатива. Используемая в нашей работе платформа публично-облачных вычислений Amazon Web Services (AWS) обладает центрами обмена данных в 15 городах мира и предоставляет клиентам вычислительные мощности в том объеме, который нужен клиенту, и исключительно тогда, когда это необходимо. Изначально облачные платформы разрабатывались для бизнес-задач, но в дальнейшем получили широкое распространение в рамках решения научных, в частности биомедицинских, исследований. В сфере омикс-технологий первый удачный опыт был получен в области протеомики, а в 2018 г публично-облачные вычисления стали основой для выравнивания геномных прочтений в STAR [17]. Облачные системы также нашли свое применение в анализе протеома [13] и микробиома [18], будучи сопряженными с конструктором конвейеров обработки данных Galaxy [19]. МЕТОДИКА
Исходные данные В предыдущем разделе были описаны общие принципы подготовки биообразцов, процедура выделения нуклеиновых кислот и условия их считывания на секвенаторе MinION. Полученные данные должны быть размещены в локальной системе хранения и депонированы в архиве Sequence Read Archive (SRA). Для разработки и тестирования протокола результаты секвенирования в виде файлов формата fast5 были загружены из системы SRA по идентификатору PRJNA635536, BioProject. Объем данных составлял 37 Гб и 102 Гб в зависимости от числа работоспособных ячеек нанопорового секвенатора MiniION. Различия в числе ячеек зависит от срока и условий хранения, а также от успешности процедуры активации, осуществляемой инжекцией реагентов в систему микроканалов чипа. Вычислительные ресурсы Для обработки результатов транскриптомного анализа, полученных с использованием технологии длинных прочтений, на облачной платформе AWS была развернута виртуальная машина с попеременно подключаемыми тремя конфигурациями условно обозначенными как t2.micro, р3.2xlarge и p2.xlarge. Конфигурации различаются по числу процессоров, объему памяти и стоимости использования (табл. 1). Конфигурация t2.micro, как наименее производительная, была использована исключительно для процедуры загрузки экспериментальных данных. Высокопроизводительную конфигурацию виртуальной машины р3.2xlarge применяли для конвертирования массива файлов формата fast5 в формат fastq с использованием перевода сигналов, полученных от нанопорового секвенатора, в последовательность нуклеотидных остатков с разметкой качества секвенирования («бейз-коллинг»). Виртуальную машину, основой которой является «средняя» конфигурация p2.xlarge, использовали для обработки данных, полученных после процедуры «бейз-коллинг», в частности, для получения статистической справки о качестве секвенирования и для расчёта полуколичественной оценки уровня экспрессии генов.
Референсный транскриптом Программное обеспечение KnowMIN, поставляемое к секвенатору MinION, генерирует набор файлов с уникальными именами в формате fast5, которые содержат информацию о считываемых транскриптах. Количество файлов определяет оператор прибора через указание предельного размера каждого записываемого на диск файла. Долговременная сохранность полученной в ходе секвенирования информации, как правило, обеспечивается на базе локальной системы хранения данных (СХД). Обьем данных производимых в результате одного запуска секвенатора, составляет от 30 Гб до 100 Гб и содержит от 50 до 150 файлов. Данные могут содержать технические и биологические повторы, что увеличивает объем необходимого дискового пространства. В протоколе мы рассматриваем последовательность шагов для обработки одного технического повтора эксперимента. Для выполнения протокола необходимо наличие файла с референсным транскриптомом человека. Референсную базу данных периодически генерируют с использованием виртуальной транскрипции генома человека. Версия указывается согласно геному; в нашей работе использовали trasncriptome.v32. Файл в формате fasta необходимо загрузить с сайта https://www.gencodegenes.org/human/ (~350 Мб). На виртуальной машине AWS файл с транскриптами генома человека размещен в директории work/ref. Каждая запись транскрипта состоит из описательной части (заголовка) и нуклеотидной последовательности. Пример заголовка записи для одного из транскриптов в базе данных Ensembl (детальное описание формата доступно в [20]):
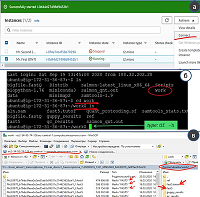
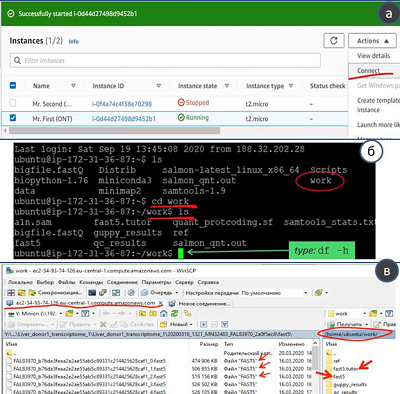
Всего база данных транскриптов человека содержит более 150 тыс. записей, относящихся к белок-кодирующим генам и их сплайс-вариантам, псевдогенам, а также генам молекул РНК, в том числе, длинным некодирующим последовательностям. Программное обеспечение и сеть На локальном компьютере пользователя должны быть установлены: современная версия веб-браузера (рекомендуется Google Chrome), а также свободно распространяемая программа-клиент, поддерживающая работу сервиса Amazon Simple Storage (Amazon S3), например, WinSCP (https://winscp.net). WinSCP используют для копирования исходных данных в формате fast5 на виртуальную машину развернутую в AWS (дополнительные материалы, приложение 1). Кроме того, должна быть установлена программа-клиент для организации удалённого доступа (например, PuTTy;https://www.putty.org/). Программа PuTTy в данном случае является эмулятором командной строки для управления виртуальной машиной AWS и проводимыми на ней вычислениями. Для установления защищенного соединения с сервером Amazon с помощью программных клиентов WinSCP и PuTTy пользователю потребуется приватный ключ-файл с расширением *.ppk (данный ключ генерируется при создании каждой виртуальной машины на площадке AWS). На удаленной виртуальной машине, расположенной в облачной системе AWS, должна быть установлена операционная система Ubuntu версии 16.4, что соответствует рекомендуемым требованиям разработчиков программного обеспечения. Для задач, рассматриваемых в этой статье, был подготовлен тестовый виртуальный сервер, условно названный Mr.First (ONT), доступ к которому можно получить у авторов по запросу, при условии соблюдения лицензионных правил компании «ONT». На сервере развернуто следующее программное обеспечение: ограниченно-распространяемая программа Guppy_basecaller, дистрибутив R с пакетом дополнительно инсталлируемых модулей и программой MinIONQC.R, а также программы Samtools и Salmon. Дополнительно загружен референсный геном (папка work\ref), а также подготовлена папка work\fast5.tutor для транскриптомных ONT-данных. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Работа с протоколом подразделяется на четыре основных этапа: (1) посредством веб-браузера необходимо зайти на сайт AWS (https://aws.amazon.com/) и включить виртуальную машину MrFirst (ONT), предварительно подключив к ней необходимую конфигурацию (стартовая конфигурация, как правило t2micro, см. Табл. 1); (2) после включения виртуальной машины, установить с ней удаленное соединение посредством программ PuTTy и WinSCP (необязательно при первичном ознакомлении) и проверить наличие доступного дискового пространства; (3) выполнить несколько команд в консольном окне программы PuTTy (дополнительные материалы, приложение 1, семь операций), скопировав их из текстового файла (дополнительные материалы, приложение 2) в рабочий терминал связи с сервером MrFirst (ONT). Необходимо проконтролировать корректное выполнение команд, сопоставив названия и размер файлов (в пределах 30% соотношение должно соответствовать значениям, приведенным в таблице 2); (4) по завершении работы, используя клиент WinSCP, скопировать полученные результаты и удалить сгенерированные файлы, выполнив последовательность команд “rm” (remove) (дополнительные материалы, приложение 2). Затем снова зайти на сайт AWS и выключить виртуальную машину, поскольку согласно расчетной политике тарифов AWS работа виртуальной машины оплачивается, в том числе, и в режиме простоя. Вышеизложенная последовательность действий пошагово приведена в дополнительных материалах (приложение 1). Для наглядной иллюстрации на рисунке 1а приведен пример окна, отображающего работу виртуальной машины с подключенной конфигурацией t2.micro. На рисунке 1б показано окно консоли программы Putty в процессе соединения с виртуальной машиной с использованием терминала. Для подключения необходимо : (1) установить в поля настройки интернет-адрес удаленной машины (предоставляется при вызове опции [Connect] из меню [Actions] (рис. 1а) и (2) указать пути к файлу ключа доступа на диске локального компьютера, служащего для авторизации при соединении с удаленным сервером. Файл ключа имеет расширение «.ppk» (предоставляется авторами статьи по запросу). При работе с терминалом необходимо построчно копировать команды, приведенные в дополнительных материалах (приложение 2). Скопировав очередную строку в буфер обмена следует перейти в командную строку PuTTy (зеленый курсор на рисунке 1б), и вставить строку из буфера обмена одним нажатием на правую кнопку мыши. Запустить команду («Enter»), дождаться завершения ее работы (появление зеленого курсора) и перейти к следующей строке-команде.
Тестовое подключение, показанное на рисунке 1б, необходимо для проверки наличия свободного дискового пространства на виртуальной машине (команда “df –h”). Если папка work заполнена более чем на 50%, то с использованием терминала необходимо удалить в папке /home/ubuntu/work ранее сгенерированные файлы с результатами расчетов (блок команд “rm” в приложении 2 дополнительных материалов). С использованием консоли можно провести проверку работоспособности описываемого конвейера обработки данных (дополнительные материалы, приложение 1) на тестовом наборе (см. папку /fast5.tutor). После этого можно приступать к копированию фaйлов, которые были получены в результате работы программы MinKNOW и являются исходными данными для выполнения протокола. На левой панели рисунка 1в представлен фрагмент окна программы WinSCP с генерируемыми MinKNOW файлами в формате fast5, каждый из которых занимает примерно 0.5 Мб. Выполнение протокола начинается с копирования исходных файлов от пользователя в директорию /fast5 на виртуальной машине. В правой панели на рисунке 1в показан пример папки на виртуальной машине AWS. Для проверки работоспособности виртуальной машины предварительно рекомендуется выполнить операции, указанные в приложении 1 дополнительных материалов, с использованием в качестве входных данных несколько файлов формата fast5, которые находятся в папке /fast5.tutor. Эта же папка может использоваться в целях обучения навыкам обработки данных с применением рассматриваемого в статье протокола. Преобразование исходных файлов, находящихся в директории work/fast5, в формат fastQ нуклеотидных последовательностей с оценкой качества прочтения каждого нуклеотида (Quality) проводится программой guppy_basecaller. С использованием веб-консоли AWS необходимо произвести замену конфигурации t2.micro используемой виртуальной машины на оснащенную GPU-процессором конфигурацию p3.2xlarge, поскольку выполнение программы без использования виртуальных GPU процессоров невозможно. Для смены конфигурации используются опции веб-консоли управления виртуальной машиной: [Instance State] > [Stop], [Change Instance Type], [Instance State] > [Start] (дополнительные материалы, приложение 1, рис. 2). На примере обработки результатов расшифровки транскриптома человека (табл. 2) можно видеть зависимость времени вычислений от объема входных данных. Наиболее времязатратным является этап работы программы Guppy_basecaller: для 37.5 Гб исходного объема входных данных в виде файлов формата fast5 вычислительное время процедуры «бейз-коллинг» составило 21 мин, а для 102 Гб – 60 мин. Возможности облачных вычислений позволяют за приемлемое время протестировать пространство вариантов транскриптома, варьируя различные параметры, контролирующие используемые алгоритмы. В таблице 2 представлен только один вариант запуска программных компонентов со стандартным набором параметров. Изменение параметров работы алгоритмов влияет на полученный спектр транскриптов: состав детектированных транскриптов в таком случае для одного набора первичных данных может изменяться на 5-7%. Значения параметров, описанные в приложении 2 дополнительных материалов, используются по умолчанию. Запуск программы Guppy_basecaller вызывает ошибку выполнения при использовании менее производительных конфигураций, чем p3.2xlarge, так как процедура «бейз-коллинг» нуждается в графическом ускорителе исключительно модели V100.
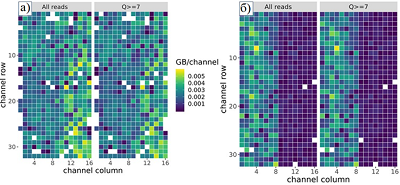
Кроме наиболее долгих по времени расчетных процедур «бейз-коллинг», «minimap2» и «salmon quant», в таблице 2 отражена менее времязатратная, но важная стадия контроля качества «бейз-коллинг» с применением программы MinIONQC.R. В качестве примера на рисунке 2 приведена сгенерированная программой MinIONQC.R тепловая диаграмма, показывающая цветом загрузку каналов секвенатора. В ячейках происходит расшифровка генетической информации, при этом пропускная способность ячейки (объем прочитанных данных, Гб) закодирована цветовой шкалой от темно-синего (минимальная) до желтого (максимальная) оттенков. Способность ячейки генерировать данные зависит от того, какое количество белковых пор в ней заработали после активации чипа. Нанопора образована достаточно сложной по структуре молекулой, встроенной в мембрану. Затянутые сроки хранения чипа, нечеткое соблюдение рекомендаций производителя и, наконец, образование в микрочипе в момент активации «воздушной пробки» в микрофлюидных каналах могут привести к выключению ячеек.
Как видно из таблицы 2, исходные массивы данных различаются примерно в 2.5 раза (37 Гб и 102 Гб). Разница в объемах данных связана с различной эффективностью работы нанопорового секвенатора, что отображено на рисунке 2 в виде тепловой карты. Если часть пор работает неэффективно (темно синий цвет, производительность менее 0.001 Гб на канал, рис. 2б), например, из-за долгосрочного хранения секвенирующего чипа, то объем получаемой информации снижается. За исключением «бейз-коллинга» остальные, перечисленные в таблице 2 этапы обработки данных, не требуют участия графического ускорителя. Из таблицы 2 следует, что целесообразно последовательное применение нескольких компьютерных конфигураций, переключение между которыми позволяет оптимизировать общее время выполнения расчетной задачи и сократить финансовые затраты, связанные с арендой различных вычислительных мощностей. После обработки данных в директории work виртуальной машины будут сохранены файлы, указанные в таблице 3 (имена файлов или директорий указаны в столбце «Результат»). Наличие и размер файлов позволяют пользователю проконтролировать успешное прохождения протокола обработки результатов секвенирования нанопорового секвенатора. Например, в папке qc_results содержатся файлы с оценкой качества исходных экспериментальных данных: - файл qc_results\summary.yaml содержит статистику по выполнению протокола: общее количество прочтений, их средняя и максимальная длина, среднее качество прочтений, информация о количестве прочтений определенной длины. - файлы qc_results\png содержат информацию в графической форме для оценки качества запуска нанопорового секвенатора: распределение прочтений по длине и по качеству, общий выход по количеству прочитанных нуклеотидных остатков, количество гигабаз, полученных с каждого канала нанопорового чипа (рис. 2). Таблица 3 содержит сведения, позволяющие планировать загрузку пользователем результатов работы к себе (например, в локальную систему хранения данных). Директория guppy_results содержащит результаты операции «бейз-коллинг», директория salmon_quant.out - результаты количественной оценки уровня экспрессии транскриптов.
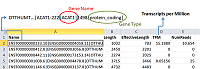
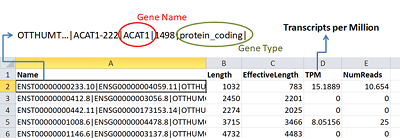
Коэффициент сжатия файлов с результатами секвенирования зависит от типа данных и способа архивации. Применение процедур архивации обусловлено сокращением времени загрузки, а также возможностью обеспечения долгосрочной защиты полученных данных при установке пароля на полученный архив. Результаты «бейз-коллинга» (папка guppy_results) могут быть сжаты в 2-3 раза (формат FastQ плохо сжимаем), тогда как размер файлов с результатами количественной оценки, большая часть которых представлена в текстовом формате, с применением пакета gzip можно уменьшить с 2 Гб до 129 Мб. Файл samtools_stats.txt содержит информацию о количестве прочтений, отображенных (картированных) на референсные транскрипты из папки work\ref, данные об общем количестве последовательностей, о количестве отфильтрованных по качеству прочтений, а также статистику, относящаяся к картированию коротких чтений. Детальное описание формата файла samtools_stats.txt доступно в [18]. Поскольку программа samtools разрабатывалась для коротких прочтений, то далеко не вся информация может оказаться применима для результатов работы нанопорового секвенатора. Текстовый файл quant_protcoding.sf (формируется с использованием команды grep, служащей для отбора строк согласно пользовательскому шаблону) содержит информацию о количестве прочтений, которые картируются на белок-кодирующие транскрипты (около 85 тыс. строк). Выгрузка файла quant_protcoding.sf на локальный компьютер осуществляется с использованием файлового менеджера WinSCP и занимает менее 10 с. После копирования файл можно просмотреть используя функцию импорта программ Excel (Microsoft) или Libre Excel (Unix); знак табуляции следует указать в качестве разделителя полей (рис. 3). В столбце «А» на рисунке 3 содержится информация об идентификаторе транскрипта и соответствующего ему гена, в столбце «B» - длина транскрипта, «С» - эффективная длина, расчет которой подробно описан в [21]. В столбце «D» приведена оценка уровня экспрессии гена в единицах «TPM» (Transcripts Per Million – число транскриптов, нормированных на миллион нуклеотидов, расшифрованных в процессе секвенирования), а в столбце «E» - количество произведённых нанопорой считываний фрагментов последовательности транскрипта.
ЗАКЛЮЧЕНИЕ В статье представлен протокол, реализующий цепочку обработки данных для количественного анализа транскриптома с применением технологии нанопорового секвенирования «ONT» и вычислительных возможностей AWS. Использование AWS оправдано, как минимум, тем, что используется Linux-ориентированное программное обеспечение, портированное адекватно только на ОС Ubuntu вер.16.4, тогда как текущей является ОС Ubuntu вер. 20.04, а аппаратная платформа тебует оснащения графическими ускорителями, которые нужны только для части вычислений. Гибкость в переключении конфигураций в облачной системе позволяет подобрать оптимальное сочетание расчетных машин с заданными конфигурациями. В перспективе схожие решения можно ожидать российских компаний Яндекс или Мейл.ру. В статье представлена инструкция (приложение 1 дополнительных материалов), цель которой - проиллюстрировать доступность сборки и анализ геномов и транскриптомов с применением нанопрового секвенатора пользователям без специальных навыков в биоинформатике. Такой подход важен для широкомасштабного вовлечения в процесс развития геномных технологий аспирантов, студентов и даже школьников для большей гибкости образовательных программ, в том числе, в условиях дистанционного обучения. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. Загруженные на сервера компании Amazon данные получены из общедоступного Интернет-источника (Sequence Read Archive). ФИНАНСИРОВАНИЕ Работа выполнена в рамках программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. БЛАГОДАРНОСТИ Авторы благодарны за плодотворное обсуждение работы сотрудникам ИБМХ: заведующей лабораторией анализа постгеномных данных Екатерине Ильгисонис и старшему научному сотруднику Ольге Киселевой. КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсустствии конфликта интересов. ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ К данной статье приложены дополнительные материалы, свободно доступные на сайте журнала (http://dx.doi.org/10.18097/BMCRM00131). ЛИТЕРАТУРА
|