|
Оценка качества прогноза анти-SARS-CoV-2 активности
Научно-исследовательский институт биомедицинской химии имени В. Н. Ореховича, Ключевые слова: SARS-CoV-2; оценка молекулярного подобия; молекулярный докинг; поиск новых антикоронавирусных соединений; тестирование качества веб-сервиса DOI: 10.18097/BMCRM00140 ВВЕДЕНИЕ
COVID-19 – передающаяся воздушно-капельным путем острая респираторная инфекция, которую вызывает представитель семейства коронавирусов, SARS-CoV-2 (2019-nCoV) [1]. По данным Всемирной организации здравоохранения, на 25 октября 2020 года в мире было выявлено около 44.8 млн. случаев заражения COVID-19 и почти 1.15 млн. летальных исходов, связанных с коронавирусной инфекцией [2]. Российская Федерация занимает 4-е место в мире по количеству заболевших с 1.51 млн. подтвержденных случаев при 17 803 955 проведенных тестах, 1.14 млн. на текущий момент разрешились выздоровлением, а 25 050 – летальным исходом [3]. По данным ReDO project, на момент написания публикации, в мире проводится 1618 клинических исследований, свыше 64% которых направлено на репозиционирование разрешенных к медицинскому применению фармакологических субстанций [4]. Такое положение, в частности, связано с тем, что репозиционирование лекарств требует меньших финансовых затрат; при этом само исследование занимает существенно более короткое время в сравнении с традиционными подходами, направленными на поиск оригинальных препаратов («first-in-the-class drugs») [5]. В настоящее время наибольшее количество исследований посвящено антибиотику азитромицину (65) и иммунодепрессанту тоцилизумабу (57). С целью поиска новых химических соединений с антикоронавирусной активностью проводится около 550 клинических испытаний [6]. Препараты, которые в настоящее время применяются для экспериментальной (off-the-label) терапии COVID-19, характеризуются умеренной эффективностью, которая зависит от стадии и тяжести течения заболевания, а также широким набором побочных эффектов, что ограничивает возможность их применения [7]. Так, результаты недавнего рандомизированного исследования эффективности ремдезивира [8] на 1062 пациентах показали, что по сравнению с плацебо данный препарат позволил добиться более быстрого выздоровления (на 5 дней), снижения смертности (на 5.2 % и 3.8% при оценке на 15-й и 29-й дни соответственно), при этом серьезные побочные эффекты наблюдались у 24.6% пациентов (впрочем, при приеме плацебо они наблюдались у 31.6% пациентов). Более того, согласно препринту группы исследователей ВОЗ [9], в котором представлены результаты рандомизированного исследования эффективности целого ряда противовирусных препаратов, применяемых в терапии COVID-19, ни один из существующих вариантов терапии не позволяет добиться существенного снижения смертности, времени госпитализации и потребности в искусственной вентиляции легких, что было показано на 11 266 пациентах, включенных в выборку случайным образом в госпиталях разных стран [8]. Таким образом, для дальнейшей разработки высокоэффективных терапевтических препаратов актуальным является поиск и создание новых фармакологических веществ, активных в отношении белков-мишеней вируса SARS-CoV-2 или белков человека, которые участвуют в патогенезе COVID-19. С целью выявления в обширных библиотеках химических соединений, перспективных для дальнейшего тестирования на антикоронавирусную активность in vitro и in vivo, широко применяют методы компьютерного конструирования лекарств. При проведении поискового запроса по ключевым словосочетаниям «COVID-19» и «Virtual screening» в Google Scholar установлено, что за десять месяцев с начала 2020 года, опубликовано свыше 42 тыс. работ, посвященных виртуальному скринингу с использованием оценки структурного сходства, методов машинного обучения и молекулярного докинга. Первым в мире свободно доступным в сети Интернет веб-ресурсом, позволяющим прогнозировать взаимодействие химических соединений с белками-мишенями, стала платформа D3Targets-2019-nCoV [10]. В её основе лежит поиск по молекулярному подобию (D3Similarity) среди 604 соединений из базы данных (БД) CoViLigands, которая была составлена разработчиками с учетом информации о химических соединениях, активных по отношению к представителям семейства коронавирусов SARS, MERS и SARS-CoV-2, и молекулярный докинг (D3Docking) к набору из 47 отобранных авторами белков-мишеней. При этом в оригинальной публикации, посвященной описанию модуля D3Similarity [10], количество соединений, содержащихся в БД CoViLigands, составляет 470. В оригинальной публикации [10], авторами приведено описание результатов тестирование модуля D3Similarity на примере двух мишеней - 3CLpro и PLPro. В ходе тестирования проводился расчёт оценок сходства между каждым активным соединением и остальными соединениями БД CoVligands. Исследователями оценивался процент верных результатов прогноза, попавших в топ 10 при ранжировании по разным оценкам сходства (2D, 3D и 2Dх3D). Установлено, что наибольший процент предсказанных верных совпадений с известными белками-мишенями наблюдается при использовании интегральной оценки 2Dх3D. Однако, как для модуля D3Similarity, так и для модуля D3Docking, независимая оценка качества прогноза, учитывающая как правильные, так и ошибочные результаты прогноза, отсутствует. В связи с этим, целью настоящей работы стало независимое проведение оценки возможности применения веб-ресурса D3Targets-2019-nCoV для отбора соединений, обладающих антикоронавирусной активностью, т.е. потенциальных фармакологических веществ для терапии SARS-CoV-2/COVID-19. МАТЕРИАЛЫ И МЕТОДЫ D3Targets-2019-nCoV Тестируемый веб-сервис предоставляет пользователям возможность проводить виртуальный скрининг с использованием двух подходов: оценки сходства структуры лигандов и молекулярного докинга к ряду белков-мишеней. Данные подходы реализованы в вычислительных модулях «D3Similarity» и «D3Docking» соответственно. Для предварительной обработки структур, как вводимых пользователем, так и содержащихся в БД, используется программа OpenBabel [11] и библиотека RDKit [12]; в результате обработки структуры химических соединений транслируются в 3D формат MOL2 и оптимизируются [13]. D3Similarity D3Similarity – вычислительный модуль платформы D3Targets-2019-nCoV для расчёта оценок сходства (2D, 3D и 2D*3D) между загруженными пользователем структурами и структурами, содержащимися в БД СoViLigands [14]. В модуле «D3Similarity» реализованы две функции: TargetPrediction и VirtualScreening. Функция TargetPrediction принимает на вход структуру одного химического соединения, представленную в формате MOL или SDF и, выводит пользователю оценку сходства со структурами соединений, содержащихся в БД CoViLigands. Пользователь получает таблицу, содержащую 20 наивысших оценок сходства. Наивысшие оценки сходства выбираются среди произведений оценок 2D и 3D сходства, так как, согласно результатам авторов [15], именно этот интегральный параметр показал наилучшую предсказательную способность. Функция VirtualScreening принимает на вход до 99 структур, однако оценка сходства проводится только с соединениями, представленными в БД CoViLigands как активные по отношению к выбранным пользователем белкам-мишеням (максимум двум). D3Docking Докинг проводится в установленные авторами потенциальные карманы связывания объёмом более 200 A3 с помощью программы AutoDock Vina [16] с последующей симуляцией молекулярной динамики моделей лиганд-белковых комплексов с применением разработанной исследователями компьютерной программы vsREMD [17]. Детальное описание подготовки структур для докинга и перечень используемых авторами компьютерных программ представлены в оригинальной публикации авторов [10]. Как и D3Similarity, модуль D3Docking включает две функции – VirtualScreening и TargetPrediction, которые позволяют проводить докинг для нескольких химических соединений (максимум 99) с одним или двумя лиганд-связывающими сайтами белка-мишени, или молекулярный докинг одного соединения со всеми доступными потенциальными карманами связывания. Тестовая выборка Тестовая выборка для проведения прогноза была составлена нами на основе информации, извлеченной из доступных источников: БД ChEMBL 27 и Coronavirus Antiviral Research Database [18, 19]. Всего в тестовую выборку вошли структуры 94 химических соединений. В тестовой выборке представлены как структуры, для которых в условиях эксперимента in vitro было продемонстрировано наличие антикоронавирусной активности по отношению к SARS-CoV-2 с указанием предполагаемых белков-мишеней, так и структуры соединений, для которых либо было показано отсутствие антикоронавирусной активности в эксперименте, либо соответствующее тестирование не проводилось. Количественные характеристики тестовой выборки с разбивкой по белкам-мишеням приведены в таблице 1.
В таблице 1 к отрицательным примерам отнесены соединения, для которых было экспериментально установлено отсутствие активности по отношению к рассматриваемым в нашей работе белкам-мишеням (16 - к 3CLpro, 7 - к PLPro, 5 -к RdRp), а также соединения, ингибирующие репликацию вируса в клеточной культуре путем воздействия на иные мишени, не представленные в БД CoViLigands (например, на ангеотензин-конвертирующий фермент (ACE2)). Структуры соединений с установленными экспериментально видами активности рассматривались в качестве положительных примеров; остальные – в качестве отрицательных примеров. Под истинно положительным (ИП) результатом прогноза понимается случай, когда прогнозируемая веб-ресурсом D3Targets-2019-nCoV для анализируемого вещества активность подтверждена экспериментально; под ложноотрицательным (ЛО) результатом прогноза понимается случай, когда активность установлена экспериментально, но не прогнозируется. В качестве истинно отрицательных (ИО) результатов нами принимались случаи, когда для неактивных соединений мишень не была предсказана; ложноположительными (ЛП) результатами считались случаи, когда для соединения прогнозировалась мишень, взаимодействие с которой экспериментально не установлено. Известно, что в большинстве случаев соединения с высоким структурным сходством проявляют сходные виды активности [20]. Поскольку ни для одного из веществ сформированной нами тестовой выборки не было исследовано взаимодействие со всеми анализируемыми в работе мишенями, мы оценили структурное сходство внутри группы соединений, активных по отношению к конкретной мишени, и их структурное сходство с соединениями, для которых взаимодействие данной мишенью не установлено. Оценку сходства проводили между химическими структурами, представленными в виде наборов двух типов подструктурных дескрипторов – MNA [21] и QNA [22]. MNA-дескрипторы (англ. Multilevel Neighbourhoods of Atoms) – представление молекулы, в котором явно указаны, согласно валентностям и зарядам атомов, все атомы водорода и не учитываются типы связей. Построение MNA-дескрипторов для каждого атома молекулы происходит рекурсивно и основано на следующем алгоритме:
QNA-дескрипторы (англ. Quantitative Neighbourhoods of Atoms) – рассчитываются с использованием значений потенциала ионизации (IP) и сродства к электрону (EA) для каждого атома молекулы. Расчёт QNA-дескрипторов производится по формулам (1-3).
Методы оценки структурного сходства на основе MNA и QNA дескрипторов детально описаны в работе [23]. Оценка точности прогноза взаимодействия анализируемых соединений с белками-мишенями COVID-19 с использованием D3Targets-2019-nCoV На основе экспериментально установленных данных о взаимодействии веществ тестовой выборки с молекулярными мишенями мы рассчитали сбалансированную точность прогноза (СТ) по формуле (4).
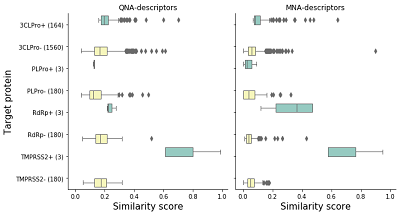
Важно заметить, что расчёт СТ проводили по совокупности полученных результатов, т. е. по всем мишеням; значения СТ для каждой из мишеней рассчитаны не были, поскольку количество активных соединений по отношению к отдельным мишеням невелико. Отметим, что авторы ресурса не приводят пороги значений сходства и оценочной функции докинга для чёткого деления анализируемых химических соединений на активные/неактивные по отношению к белкам-мишеням. Поэтому, в процессе работы, мы использовали результаты оценки сходства и расчёта оценочной функции докинга, полученные для соединений тестовой выборки и белков-мишеней, активность в отношении которых подтверждена в условиях тестирования in vitro. Это соответствует подходу, использованному авторами веб-ресурса [10]. РЕЗУЛЬТАТЫ Сравнение оценок сходства соединений тестовой выборки Оценки сходства для активных и отрицательных примеров соединений и их сопоставление приведены на рисунке 1.
На рисунке 1 распределения оценок сходства для каждой из групп представлены в виде прямоугольников. Левая граница прямоугольника указывает на значение нижнего квантиля, правая – значение верхнего квантиля. Границы, выходящие на пределы прямоугольника, соответствуют минимальному и максимальному значению из совокупности оценок сходства внутри каждой из групп. Выбросы обозначены в виде отдельных точек. Вертикальная линия, локализованная внутри прямоугольника, указывает на значение медианы. Как видно из приведённых на рисунке 1 данных, оценки сходства внутри группы соединений, активных по отношению к TMPRSS2 (мембрано-связанная сериновая протеаза, единственная мишень, представляющая белок человека), существенно выше, чем оценки сходства активных соединений с отрицательными примерами для TMPRSS2 (3 протестированных соединения и 59 отрицательных примеров). Такие результаты получены как при оценке сходства по MNA-дескрипторам (среднее значение для активных соединений равно 0.70±0.18, среднее значение оценок сходства отрицательных примеров с активными соединениями равно 0.06±0.04), так и при оценке сходства по QNA-дескрипторам (0.74±0.18 для группы активных соединений, 0.17±0.06 для оценки сходства между активными соединениями и отрицательными примерами). Для группы активных соединений и оценки сходства отрицательных примеров и активными соединениями в отношении РНК-зависимой РНК-полимеразы RdRp получен аналогичный результат. Среднее значение полученных оценок сходства по MNA-дескрипторам для группы активных соединений равно 0.33±0.15, по QNA-дескрипторам – 0.24±0.03. Для оценки сходства между отрицательными примерами и активными соединениями среднее значение оценки сходства по MNA-дескрипторам равно 0.05±0.04, по QNA-дескрипторам – 0.18±0.06. В анализируемой нами тестовой выборке только три соединения обладают активностью по отношению к PLpro. Значение оценки сходства по MNA-дескрипторам составляет 0.04±0.03, по QNA-дескрипторам составляет 0.13±0.01. Среднее значение оценки сходства между отрицательными примерами и соединениями, активными в отношении PLpro, по MNA-дескрипторам составляет 0.05±0.04, по QNA-дескрипторам составляет 0.14±0.07. Средняя оценка сходства для соединений, активных по отношению 3CLpro, по MNA-дескрипторам равна 0.12±0.08, по QNA-дескрипторам равна 0.22±0.07. Для отрицательных примеров и соединений, активных по отношению к 3CLpro, средние оценки составляют 0.06±0.05 по MNA-дескрипторам и 0.18±0.07 по QNA-дескрипторам. Таким образом, значимые структурные различия наблюдаются для TMPRSS2, и RdRp. В случае PLpro значимых различий по сходству активных соединений друг с другом и с отрицательными примерами не наблюдается, а для 3CLpro активные соединения также незначительно отличаются от отрицательных примеров. Оценка применения методов подобия, реализованных в модуле D3Similarity Возможности использования функции TargetPrediction модуля D3Similarity мы рассмотрели на примере прогноза для структурной формулы препарата тидеглусиб. Тидеглусиб – селективный ингибитор киназы гликогенсинтазы 3 бета (GSK3-beta), который в настоящее время проходит клинические исследования как препарат для лечения болезни Альцгеймера [24]. В исследованиях in vitro было показано, что тидеглусиб является ингибитором основной протеазы вируса SARS-CoV-2 (3C-like protease, 3CLpro) [25]. Результаты оценки его сходства со структурными формулами веществ, содержащихся в БД CoViLigands, приведены в таблице 2.
Таблицы 2-5, содержащие результаты расчёта оценочной функции докинга и оценки сходства, приведены в соответствии с представлением на тестируемом веб-сервисе. Название столбца «Target ID» заменено на «UniProt ID/ProteinID». Как видно из приведенных в таблице 2 данных, тидеглусиб содержится в базе данных CoViLigands (ICV265), и оценка 2D сходства позволяет его идентифицировать (сходство равно 100%). Ранее было отмечено, что структуры, представленные в БД CoViLigands, и структуры, загруженные пользователем, подвергаются идентичной предобработке. При этом, однако, оценка 3D сходства между двумя структурами тидеглусиба составляет только 78.90%. Было также обнаружено сходство с другими ингибиторами основной протеазы (ICV189, ICV197 и ICV193); для них оценки 2D сходства составляют 20.88%, 22.54% и 19.28%, а 3D сходства 79.90%, 73.95% и 80.71%, соответственно. Помимо вышеупомянутых 4-х ингибиторов основной протеазы SARS-CoV-2, в БД CoViLigands содержатся 266 соединений с аналогичной активностью, оценка сходства с которыми для тидеглусиба ниже, чем для соединений, вошедших в список Top 20 (табл. 2). Необходимо отметить, что в Top 20 вошли мишени, о взаимодействии с которыми препарата тидеглусиб сведения отсутствуют (Histamine H1 receptor, Histamine N-methyltransferase и Papain-like protease), а также 3 неизвестные мишени (в табл. 2 Unknown). Опираясь на сведения, представленные в ячейке «Activity», можно сделать вывод о том, что Unknown, вероятно, указывает на наличие антикоронавирусной активности, установленной при тестировании в культуре клеток без идентификации молекулярной мишени. Результаты работы функции VirtualScreening модуля D3Similarity были получены с использованием SD файла (SDF, Structure Data File), содержащего все структуры тестовой выборки (табл. 1). Оценки сходства, полученные с использованием функции VirtualScreening, приведены в таблице 3. Как видно из приведенных в таблице 3 данных, для всех загруженных нами соединений, активных по отношению к 3CLpro и PLpro, оценки 2D сходства составляют 100%; следовательно, их структуры представлены в БД CoViLigands. Как уже говорилось выше, загруженные для оценки сходства и представленные в БД CoViLigands соединения проходят одинаковые процедуры предобработки и приводятся к единому формату. Из таблицы 3, однако, видно, что оценки 3D сходства для идентичных соединений не достигают 100%. Также было установлено, что в полученных результатах многократно встречаются случаи, когда оценка 3D сходства между идентичными соединениями превышает оценку 3D сходства соединений самих с собой. Примеры таких случаев представлены в таблице 4. В связи с этим возникает вопрос о корректности методов оценки 3D сходства между анализируемыми и представленными в БД структурами.
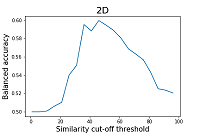
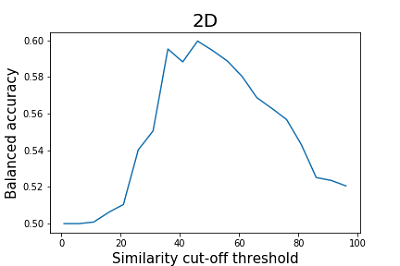
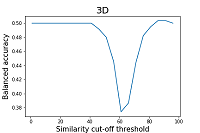
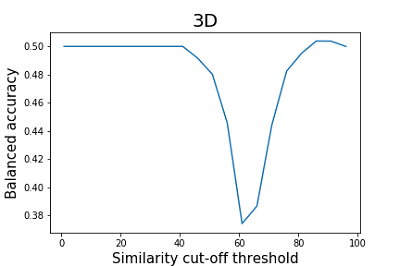
Функции TargetPrediction и VirtualScreening модуля D3Similarity основаны на идентичных алгоритмах, однако TargetPrediction позволяет получить результаты для интерпретации сразу по многим мишеням; поэтому в дальнейшей работе по оценке качества прогноза, мы использовали именно функцию TargetPrediction. Поскольку авторами работы не указан порог значений сходства для разделения соединений на активные и неактивные, нами была произведена оценка сбалансированной точности при разных порогах отсечения для оценок сходства двумерных и трехмерных структур. При этом результаты прогноза, превышающие установленный порог, считались положительными, а результаты, находящиеся ниже порога – отрицательны ми. Результаты оценки приведены на рисунках 2 и 3.
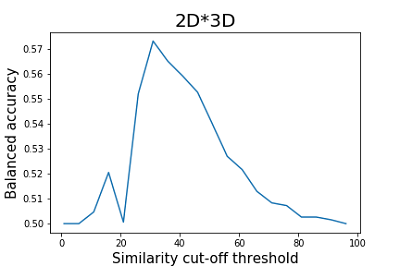
Как видно из приведенных на рисунках 2 и 3 данных, результаты расчёта сбалансированной точности для 2D и 3D методов существенно отличаются. Наибольшее значение сбалансированной точности (0.59) для 2D методов наблюдается при пороге отсечения по сходству равному 46%. Для 3D методов значения сбалансированной точности не превышает 0.51. Минимальное значение сбалансированной точности для 3D методов достигается при пороге отсечения по сходству 61 (0.37), что соответствует значениям чувствительности 0.53 и специфичности 0.22. Параметром, который рекомендуется авторами работы для оценки сходства двух структур является Similarity (2Dх3D) [15]. По мнению авторов, эта операция, помогает сгладить влияние недостатков отдельных методов, тем самым повышая качество итоговых оценок. Результаты расчёта сбалансированной точности для данного параметра приведены на рисунке 4.
Как видно из представленных на рисунке 4 данных, максимальное значение сбалансированной точности (0.57) достигается при пороге отсечения по сходству, равному 31%, что ниже, чем таковая для 2D методов. Как описано выше, при проведении оценки качества прогноза авторами работы [10] учитывались только положительные примеры результатов. Однако с использованием отрицательных примеров нами показано, что применение интегральной оценки сходства (2Dх3D) на использованной нами тестовой выборке не является информативным, а с учётом приведенных выше замечаний о некорректности оценки сходства 3D методами, может вызвать затруднения при интерпретации полученных результатов у пользователей ресурса. Оценка результатов молекулярного докинга, полученных с помощью D3Docking Для демонстрации результатов работы функции VirtualScreening модуля D3Docking был использован SD файл, в который были включены все структуры тестовой выборки (табл. 1). Результаты докинга соединений тестовой выборки к папаин-подобной протеазе и главной протеазе SARS-CoV-2 представлены в таблицах 5 и 6 соответственно.
Как видно из представленных в таблице 5 данных, для соединения GRL0617 значение оценочной функции докинга к папаин-подобной протеазе (PLpro) равно -10.49, что является наилучшим результатом среди всех оценок. Для соединения 6-тиогаунин значение оценочной функции составило -6.06. Такое значение является наихудшим среди всех расчетных значений оценочной функции докинга соединений тестовой выборки к PLpro.
Ранее мы отмечали, что активными по отношению к основной протеазе являются соединения GC-376, Тидеглусиб, C25H22FN4O4 и CHEMBL1388469. Значения оценочной функции для данных соединений составляют -8.14954, -8.25794, -9.90902, -7.64298 соответственно. Среднее значение оценочной функции, рассчитанное для соединений, потенциально неактивных в отношении основной протеазы, равно -8.48±0.8. Таким образом, C25H22FN4O4 имеет значение оценочной функции, существенно превышающее среднее значение, а для остальных соединений значение оценочной функции ниже среднего. Только для двух соединений (ремдесивир и CHEMBL1809259) расчетные значения оценочной функции существенно превышают средние значения оценочной функции для неактивных соединений – -9.49 и -9.56 соответственно. Результаты, полученные с использованием функции TargetPrediction модуля D3Docking, для препарата тидеглусиб представлены в таблице 7.
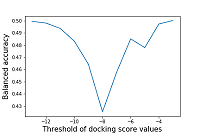
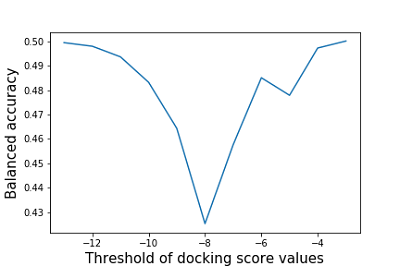
Порог значений оценочной функции докинга, опираясь на который, согласно прогнозу, можно было бы разделить соединения на активные/неактивные по отношению к белкам-мишеням, авторами не указан. В настоящей работе в качестве такого порога нами использованы значения оценочной функции, полученные при докинге лигандов к белкам-мишеням, активность в отношении которых подтверждена в условиях in vitro. Расчет среднего значения оценочной функции для 45 белков-мишеней, в отношении которых активность тидеглусиба не подтверждена экспериментально, на основе представленных в таблице 5 данных даёт величину -8.24±1.00. Вышеуказанные расчетные значения оценочной функции для двух белков-мишеней (GSK3-beta и 3CLpro), в отношении которых для препарата тидеглусиб была установлена активность, близки к данному порогу, либо ниже его. Результаты расчёта оценочной функции докинга препарата тидеглусиб с бета киназой гликоген-синтазы 3(GSK3-beta) равны -8.84, а с главной протеазой (3CLpro) варьируют в диапазоне от -8.31 до -7.35 в зависимости от выбранной трехмерной структуры белка-мишени. Наилучшие значения оценочной функции получены для дигидрооротат дегидрогеназы (DHODH) – -10.65, взаимодействие с которой препарата тидеглусиб экспериментально не установлено. Для 17 белков-мишеней значения оценочной функции докинга препарата тидеглусиб не уступают таковым, полученным для главной протеазы (-8.84). Результаты расчёта оценочной функции докинга тидеглусиба к белкам-мишеням DHODH, AKT1 и PDE5 равны -10.65, -10.57 и -10.21 соответственно. Полученные значения оценочной функции являются наилучшими даже среди тех белков-мишеней, активность тидеглусиба в отношении которых подтверждена в условиях тестирования in vitro. Вероятно, такого рода информация может быть использована для дальнейшего экспериментального исследования действия данного препарата на эти белки-мишени. Оценка сбалансированной точности используемых авторами подходов рассчитывалась на основе значений оценочной функции докинга. Результаты изменения чувствительности в зависимости от выбранного порога отсечения по значению оценочной функции приведены на рисунке 5.
Как видно из приведенных на рисунке 5 данных, значения сбалансированной точности не превышают 0.5. Минимальное значение сбалансированной точности достигается при пороге значений оценочной функции равном -8 (0.42), что соответствует значениям чувствительности 0.24 и специфичности 0.60. ЗАКЛЮЧЕНИЕ В связи с остротой проблемы пандемии коронавирусной инфекции SARS-CoV-2/COVID-19 и отсутствием достаточно эффективных и безопасных терапевтических средств, поиск новых фармакологических веществ с антикоронавирусной активностью крайне актуален [1]. Первым свободно доступным в сети Интернет веб-ресурсом, который позволяет проводить оценку сходства и молекулярный докинг для выявления соединений, потенциально пригодных для терапии COVID-19, стала платформа D3Targets-2019-nCoV. В данной работе мы оценили качество прогноза с помощью данной платформы для 94 веществ тестовой выборки, содержащей 35 активных соединений и 59 соединений, которые включают в себя соединения с установленным отсутствием активности в отношении выбранных мишеней и соединения, активные по отношению к мишеням, не представленным в БД CoViLigands. Нами была проведена оценка достоверности прогноза с использованием параметра, рекомендованного авторами, а именно произведением оценок двумерного и трехмерного сходства (2Dх3D). В ходе оценки качества прогноза показано, что максимум сбалансированной точности (0.57) достигается при пороге отсечения по сходству, равному 31%. При расчёте сбалансированной точности молекулярного докинга показано, что её значения не превышают 0.5. Помимо этого, было установлено, что максимальное значение сбалансированной точности 2D метода оценки сходства (0.59) наблюдаются при пороге отсечения по сходству, равном 46%. Для метода оценки 3D сходства максимальное значение сбалансированной точности не превышает 0.51. При анализе результатов, полученных при работе с модулем D3Similarity установлено, что для идентичных структур оценка 2D сходства составляет 100%, однако, при этом оценка 3D сходства ниже. Принимая во внимание тот факт, что загруженные нами для анализа и представленные в БД CoViLigands структуры соединений, проходят одинаковую предобработку, возникает вопрос о корректности выбранных авторами веб-сервиса методов для анализа сходства трехмерных структур. Исходя из значений сбалансированной точности для всего спектра прогнозируемых мишеней, полученных на сформированной нами тестовой выборке, и установленной несогласованности результатов расчёта структурного сходства можно сделать вывод об ограниченной полезности прогноза веб-сервиса D3Target-2019-nCoV. Таким образом, данный ресурс требует серьёзной доработки, включающей в себя в том числе и усовершенствование используемых алгоритмов, в частности методов оценки 3D сходства. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. ФИНАНСИРОВАНИЕ Работа выполнена при поддержке проекта РФФИ № 20-04-60285. КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ К данной статье приложены дополнительные материалы, свободно доступные на сайте журнала (http://dx.doi.org/10.18097/BMCRM00140). ЛИТЕРАТУРА
|