|
Assessing the Prediction Quality of the Anti-SARS-CoV-2 Activity Institute of Biomedical Chemistry, 10 Pogodinskaya str., Moscow, 119121 Russia ;*e-mail: ionov.nikita.serg@gmail.com Key words: SARS-CoV-2; estimation of molecular similarity; molecular docking; finding of new anticoronaviral agents; estimating of the web-service quality DOI: 10.18097/BMCRM00140 The D3Targets-2019-nCoV web service predicting the interaction of chemical compounds with SARS-CoV-2 virus proteins and human proteins involved in the pathogenesis of COVID-19 by structural similarity and molecular docking was evaluated. The quality of the prediction was assessed as a balanced accuracy, which was calculated based on the results of the prediction for the structures of chemical compounds from the test set we compiled. The test set consisted of 35 active and 59 inactive molecules, including compounds with the experimetnaly confrmed absence of activity against the selected targets and compounds active against SARS-CoV-2 targets, not presented in the CoViLigands database. The authors of the analyzed web service did not indicate the thresholds for the values of the similarity score and the docking scoring function, using which it would be possible to reliably divide the compounds into active and inactive with respect to target proteins. Therefore, we assessed the balanced accuracy of the predictive methods D3Targets-2019-nCoV at various thresholds for cutting off active substances from inactive ones. Using our test set it was found that the highest value of balanced accuracy (0.59) was achieved when choosing active molecules based on the results of 2D similarity assessment (cutoff threshold was 46%). Assessment of 3D similarity did not allow achieving balanced accuracy values exceeding 0.5. It is shown that using the 2Dх3D integral similarity assessment recommended by the authors, the maximum value of the balanced accuracy 0.57 was achieved at a threshold of 31%. The calculated balanced accuracy for molecular docking results does not exceed 0.51. On the case study for the tideglusib, it was shown that the values of the scoring function for two target proteins, the activity against which was confrmed in the experiment (3CLpro and GSK3B), do not differ signifcantly from the values of the scoring function for the remaining 44 targets were not confrmed. INTRODUCTION
COVID-19 is an acute respiratory infection transmitted by airborne droplets caused by a coronavirus family member, SARS-CoV-2 (2019-nCoV) [1]. According to the World Health Organization, as of October 25, 2020, about 44.8 million cases of COVID-19 infection and almost 1.15 million deaths associated with coronavirus infection were detected in the world [2]. The Russian Federation has the fourth-highest number of cases globally, with 1.51 million confirmed cases with 17803955 tests performed, 1.14 million have now been resolved, and 25050 are fatal [3]. According to the ReDO project, at the time of writing, 1618 clinical trials are being conducted worldwide, over 64% of which aim to reposition approved pharmacological substances for medical use [4]. This situation, in particular, is connected with the fact that repositioning of drugs requires less financial expenses; at the same time, the research itself takes a significantly shorter time in comparison with traditional approaches aimed at the search for original drugs ("first-in-the-class drugs") [5]. The biggest number of studies is currently devoted to the antibiotic azithromycin (65) and the immunosuppressant tocilizumab (57). Nevertheless, there are about 550 clinical trials to find new chemical compounds with anticoronavirus activity [6]. The drugs that are currently used for experimental (off-the-label) therapy of COVID-19 are characterized by moderate efficacy, the value of which depends on the stage and severity of the course of the disease, as well as a wide range of side effects, which limits the possibility of their use [7]. Thus, the results of a recent randomized study of the efficacy of remdesivir [8] on 1062 patients showed that, compared with placebo, this drug made it possible to achieve a faster recovery (by five days), a decrease in mortality (by 5.2% and 3.8% when assessed at the 15th and 29 days, respectively), while serious side effects were observed in 24.6% of patients (however, when taking placebo, they were observed in 31.6% of patients). Moreover, according to the preprint of the WHO research group [9], which presents the results of a randomized study of the effectiveness of several antiviral drugs used in the treatment of COVID-19; none of the existing treatment options can achieve a significant reduction in mortality, hospitalization time and the need for mechanical ventilation, as was shown in 11 266 patients randomly enrolled in hospitals in different countries [8]. Thus, for the further development of highly effective therapeutic drugs, it is urgent to search for and create new pharmacological substances that are active against target proteins of the SARS-CoV-2 virus or human proteins involved in the pathogenesis of COVID-19. To identify chemical compounds in vast libraries promising for further testing for anti-coronavirus activity in vitro and in vivo, computer-aided drug design methods are widely used. When conducting a search query for the keywords "COVID-19" and "Virtual screening" in Google Scholar, what found that over ten months from the beginning of 2020, more than 42 thousand scientific papers were published on virtual screening using structural similarity assessment, machine training, and molecular docking. The D3Targets-2019-nCoV platform [10] was the world's first freely available web resource that allows predicting the interaction of chemical compounds with target proteins. It is based on molecular similarity search (D3Similarity) among 604 compounds from the CoViLigands database (DB), which was compiled by the developers taking into account the information on chemical compounds active against members of the coronavirus family SARS, MERS, and SARS-CoV-2, and molecular docking (D3Docking) to the set of 47 target proteins selected by the authors. Meanwhile, in the original publication describing the D3Similarity module [10], the number of compounds in the CoViLigands database is 470. In the original publication, the authors described the D3Similarity module testing using two targets, 3CLpro and PLPro. During testing, what calculated similarity estimates between each active compound and the rest of the CoVligands database. Researchers evaluated the percentage of correct prediction results in the top 10 when ranked by different similarity scores (2D, 3D, and 2Dх3D). It was found that the highest percentage of predicted correct matches with known target proteins was observed using the 2Dх3D integral score. However, for both the D3Similarity module and the D3Docking module, there is no independent estimate of prediction quality that considers both correct and incorrect prediction results. Therefore, the present study aimed to independently evaluate the feasibility of using the web resource D3Targets-2019-nCoV to select compounds with anticoronavirus activity as potential pharmacological agents for SARS-CoV-2/COVID-19 therapy. MATERIALS AND METHODS
D3Targets-2019-nCoV The web service under test enables users to perform virtual screening using two approaches: ligand structure similarity assessment and molecular docking to several target proteins. These approaches are implemented in the computational modules "D3Similarity" and "D3Docking", respectively. The OpenBabel program [11] and RDKit library [12] are used for preprocessing the structures, both user-entered and contained in the database; as a result of processing, the structures of chemical compounds are translated into 3D format MOL2 and optimized [13]. D3Similarity D3Similarity is a computational module of the D3Targets-2019-nCoV platform for calculating similarity estimates (2D, 3D, and 2D*3D) between user-loaded structures and structures contained in the CoViLigands database [14]. The "D3Similarity" module implements two functions: TargetPrediction and VirtualScreening. The TargetPrediction function takes as input the structure of one chemical compound represented in MOL or SDF format and output to the user a similarity score with the compounds' structures in the CoViLigands database. Specifically: the user receives a table containing the 20 highest similarity scores. The highest similarity scores are chosen among the products of 2D and 3D similarity scores as an integral parameter that showed the best predictive ability, according to the authors [15]. The VirtualScreening function accepts up to 99 structures as input. The similarity assessment is still performed only with compounds presented in the CoViLigands database as active against user-selected target proteins (a maximum of two). For more details on 2D and 3D similarity estimation methods, see the original publication [15]. D3Docking Docking is performed in the potential binding pockets of more than 200 A3, established by the authors, using the AutoDock Vina program [16], followed by simulation of the molecular dynamics of ligand-protein complex models using the vsREMD computer program developed by the researchers [17]. A detailed description of the preparation of structures for docking and the authors' list of computer programs is presented in the original publication [10]. Like D3Similarity, the D3Docking module includes two functions, VirtualScreening and TargetPrediction, which allow docking for multiple chemical compounds (maximum 99) with one or two ligand-binding sites of the target protein, or molecular docking of a single compound with all potential binding pockets available. Test Set We compiled the test set based on information extracted from available sources: the ChEMBL 27 database and the Coronavirus Antiviral Research Database, curated by Stanford University [18, 19]. A total of 94 chemical compound structures were included in the test set. The test set includes the structures for which anticoronaviral activity against SARS-CoV-2 was demonstrated under in vitro experimental conditions with the indication of the putative target proteins. Also, the structures of compounds for which either the absence of anticoronavirus activity was shown in the experiment or the corresponding testing was not performed were included. Quantitative characteristics of the test set by target proteins are shown in Table 1.
In Table 1, negative examples include compounds that were experimentally found to have no activity against the target proteins considered in our work (16 to 3CLpro, 7 to PLPro, 5 to RdRp), as well as compounds that inhibit virus replication in cell culture by acting on other targets not represented in the CoViLigands database (for example, on ACE2).
Methods for assessing structural similarity based on MNA and QNA descriptors are described in detail in [23]. Evaluation of the accuracy of the prediction of the interaction of analyzed compounds with COVID-19 target proteins using D3Targets-2019-nCoV Based on the experimentally established data on the interaction of the test set substances with molecular targets, we calculated the balanced prediction accuracy (BA) using formula (4).
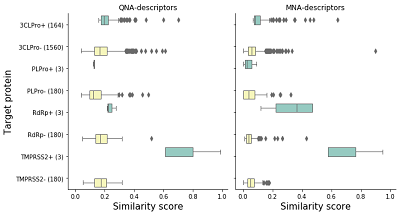
It is important to note that the BA was calculated using the totality of the results obtained, i.e., for all targets; the BA values for each target were not calculated separately. This approach was used because the number of active compounds relative to individual targets is small. Note that the resource authors do not provide thresholds for the similarity values and the estimated scoring function to divide the analyzed chemical compounds into active/inactive to the target proteins. Therefore, in the course of this work, we used the results of the similarity assessment and the calculation of the estimated scoring function obtained for the compounds of the test set and the target proteins whose activity for which has been confirmed under in vitro testing conditions. This corresponds to the approach used by the authors of the web resource [10]. RESULTS AND DISCUSSION Comparison of similarity scores of test set compounds Similarity estimates for the active and negative example compounds and their comparisons are shown in Figure 1.
Figure 1 shows the distribution of similarity scores for each group as rectangles. The rectangle's left border indicates the lower quantile value, while the right border indicates the value of the upper quantile. Borders outside the rectangle correspond to the minimum and maximum values of the set of similarity scores within each group. Outliers are marked as individual points. The vertical line localized inside the rectangle indicates the value of the median. As can be seen from the data shown in Figure 1, the similarity scores within the group of compounds active against TMPRSS2 (the only human protein target among those evaluated) are significantly higher than the similarity scores of active compounds with negative examples for TMPRSS2 (3 tested compounds and 59 negative examples). Such results were obtained both in the MNA descriptor similarity assessment (mean value for active compounds is 0.70±0.18, mean value of similarity estimates of negative examples with active compounds is 0.06±0.04) and in the QNA descriptor similarity assessment (0.74±0.18 for the active compound group, 0.17±0.06 for similarity estimates between active compounds and negative examples). A similar result was obtained for active compounds and the similarity assessment for the negative examples and active compounds for RNA-dependent RNA polymerase RdRp. The average value of obtained similarity estimations with MNA descriptors for the group of active compounds was 0.33±0.15, and with QNA descriptors - 0.24±0.03. The similarity estimation between negative examples and active compounds the average value of MNA descriptor similarity was 0.05±0.04, with QNA descriptor - 0.18±0.06. In the test set, we analyzed, only three compounds have activity against PLpro. The similarity score for MNA descriptors is 0.04±0.03, for QNA descriptors is 0.13±0.01. The average similarity score between the negative examples and the compounds active against PLpro is 0.05±0.04 for MNA descriptors, for QNA descriptors is 0.14±0.07. The mean similarity score for compounds active against 3CLpro is 0.12±0.08 for MNA descriptors and 0.22±0.07 for QNA descriptors. For negative examples and compounds active against 3CLpro, the mean scores are 0.06±0.05 for MNA descriptors and 0.18±0.07 for QNA descriptors. Thus, significant structural differences are observed for TMPRSS2 and RdRp. In the case of PLpro, no significant differences in the similarity of the active compounds with each other and the negative examples are observed. For 3CLpro, the active compounds also do not have a significant difference from the negative examples. Evaluation of the application of similarity methods implemented in the D3Similarity module We examined the possibilities of using the TargetPrediction function of the D3Similarity module on the example of prediction for the structural formula of the drug tideglusib. Tideglusib is a selective inhibitor of glycogen synthase kinase 3-beta (GSK3-beta), currently under clinical trials as a drug for treating Alzheimer's disease [24]. In in vitro studies, tideglusib is an inhibitor of the main protease of the SARS-CoV-2 virus (3C-like protease, 3CLpro) [25]. The results of its similarity assessment to the CoViLigands database's substances' structural formulas are shown in Table 2.
Tables 2-5, containing the results of calculating the estimated scoring function and the estimated function, are given according to the web service's representation under test. The name of the column "Target ID" has been changed to "UniProt ID/ProteinID". As can be seen from the data shown in Table 2, tideglusib is contained in the CoViLigands database (ICV265), and the 2D similarity assessment allows its identification (similarity equals 100%). It was noted earlier that the structures presented in the CoViLigands database and the structures loaded by the user undergo identical preprocessing. However, the 3D similarity score between the two tideglusib structures is only 78.90%. Similarities were also found with the other major protease inhibitors (ICV189, ICV197, and ICV193); for these, the 2D similarity estimates are 20.88%, 22.54%, 19.28%, and the 3D similarity estimates are 79.90%, 73.95%, and 80.71%, respectively. In addition to those mentioned above 4 SARS-CoV-2 major protease inhibitors, the CoViLigands database contains 266 compounds with similar activity, whose similarity scores for tideglusib are lower than those for the Top 20 compounds (Table 2). One should note that the Top 20 includes targets with no information on the interaction of the tideglusib drug (Histamine H1 receptor, Histamine N-methyltransferase, and Papain-like protease), as well as three unknown targets (Table 2). Based on the information provided in the Activity cell, we can conclude that Unknown probably indicates anticoronavirus activity established when tested in cell culture without identifying the molecular target. The results of the VirtualScreening function of the D3Similarity module were obtained using an SD file (SDF, Structure Data File), which contains all test set structures (Table 1). Similarity estimates obtained using the VirtualScreening function are shown in Table 3. As can be seen from the data shown in Table 3, for all of our loaded connections active to 3CLpro and PLpro, the 2D similarity scores are 100%; hence, their structures are represented in the CoViLigands database. As mentioned above, connections loaded for similarity estimation and presented in the CoViLigands database go through the same preprocessing procedures and are reduced to the same format. Table 3, however, shows that the 3D similarity estimates for identical compounds do not reach 100%. It was also found that many times in the results obtained, there are cases where the 3D similarity score between identical compounds exceeds the 3D similarity score of the compounds themselves. Examples of such cases are presented in Table 4. This raises the question regarding the correctness of the methods for estimating the 3D similarity between the analyzed and the structures represented in the database.
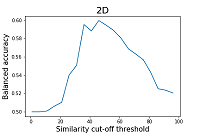
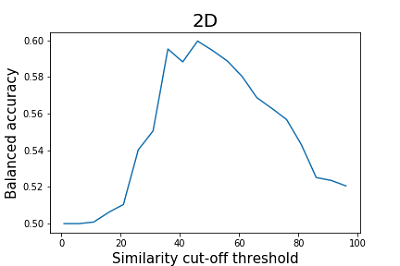
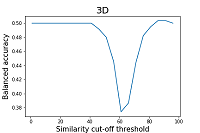
The TargetPrediction and VirtualScreening functions of the D3Similarity module are based on identical algorithms, but TargetPrediction allows you to get results for interpretation on many targets at once; therefore, in further work on prediction quality assessment, we used the TargetPrediction function. Since the authors of the paper did not specify a threshold of similarity values for separating compounds into active and inactive ones, we evaluated the balanced accuracy at different cutoff thresholds for estimating the similarity of two- and three-dimensional structures. With this approach, prediction results above the threshold were considered positive, while those below the threshold were considered negative. The estimation results are shown in Figures 2 and 3.
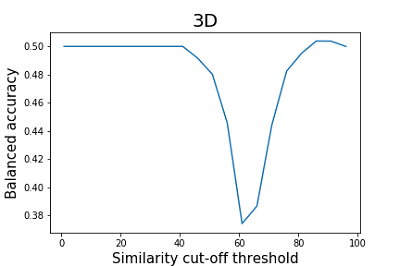
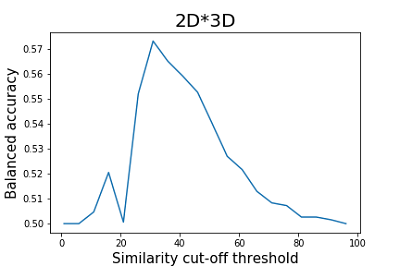
As can be seen from the data shown in Figures 2 and 3, the results of the balanced accuracy calculation for 2D and 3D methods differ significantly. The highest value of balanced accuracy (0.59) for 2D methods is observed at the similarity cutoff threshold of 46%. For 3D methods, balanced accuracy values do not exceed 0.51. A minimal value of balanced accuracy for 3D methods is reached at a similarity cutoff threshold of 61 (0.37), which corresponds to a sensitivity of 0.53 and specificity of 0.22. The parameter that is recommended by the authors of the work to assess the similarity of the two structures is Similarity (2D*3D) [15]. According to the authors, this operation helps to smooth the influence of particular methods' shortcomings, thereby improving the final results' quality. The results of the calculation of balanced accuracy for this parameter are shown in Figure 4.
As can be seen from the data presented in Figure 4, the maximum value of balanced accuracy (0.57) is achieved at a similarity cutoff threshold of 31%, which is lower than that for 2D methods. As described earlier, the authors considered only positive examples of the results when evaluating the prediction quality. However, using negative examples, we show that the application of the integral similarity assessment (2D*3D) on our test set is not informative, and given the above remarks about the incorrectness of similarity assessment by 3D methods, can cause difficulties in interpreting the results by the users of the resource. Evaluation of molecular docking results obtained with D3Docking To demonstrate the results of the VirtualScreening function of the D3Docking module, an SD file was used that included all structures of the test set (Table 1). The results of docking the test set compounds to the papain-like protease and the main protease SARS-CoV-2 are presented in Tables 5 and 6, respectively.
As can be seen from the data presented in Table 5, for compound GRL0617, the value of the estimated scoring function to papain-like protease (PLpro) is -10.49, which is the best result among all evaluations. For compound 6-thioguanine, the value of the estimated scoring function is -6.06. This value is the worst among all estimated values of the test set compounds' estimated scoring function to PLpro.
We noted earlier that the compounds GC-376, tideglusib, C25H22FN4O4, and CHEMBL1388469 are active to the main protease. These compounds' estimated function values are -8.14954, -8.25794, -9.90902, and -7.64298, respectively. The mean value of the estimated function calculated for the compounds potentially inactive against the major protease is -8.48±0.8. Thus, C25H22FN4O4 has an estimated function value that is significantly higher than the mean value, while for the remaining compounds, the estimated function value is below the mean. Only for two compounds (remdesivir and CHEMBL1809259), the estimated function's calculated values significantly exceed the average values of the estimated function for inactive compounds -9.49 and -9.56, respectively. The results obtained using the TargetPrediction function of the D3Docking module for the drug tideglusib are presented in Table 7.
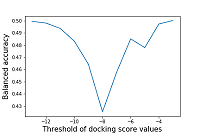
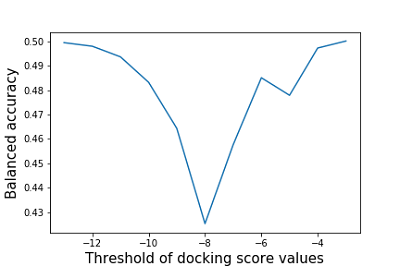
The authors did not specify the estimated scoring function threshold, based on which one could divide the compounds into active/inactive to the target proteins [10]. In the present work, we use a threshold of the estimated function values obtained by docking ligands to target proteins whose activity to them has been confirmed under in vitro conditions. Calculation of the mean value of the estimated function for 45 target proteins against which tideglusib activity has not been experimentally confirmed based on the data presented in Table 5 gives -8.24±1.00. The above-calculated values of the estimated function for the two target proteins (GSK3-beta and 3CLpro) for which tideglusib was active are close to this threshold or lower. The tideglusib results with glycogen synthase beta kinase 3 (GSK3-beta) estimated scoring function were -8.84. The main protease (3CLpro) scoring function ranged from -8.31 to -7.35 depending on the target protein's selected three-dimensional structure. The estimated scoring function's best values were obtained for dihydroorotate dehydrogenase (DHODH) -10.65, the interaction of tideglusib with which was not experimentally established. For the 17 target proteins, the tideglusib's estimated scoring function values are not inferior to those obtained for the main protease (-8.84). Calculating tideglusib's estimated scoring function to the target proteins DHODH, AKT1 and PDE5 are -10.65, -10.57, and -10.21, respectively. The estimated function's obtained values are the best even among those target proteins against which tideglusib's activity was confirmed under in vitro testing conditions. This kind of information can probably be used for further experimental investigation of this drug's action on these target proteins. The authors' estimated balanced accuracy of the approaches used was calculated based on the estimated scoring function's values. The results of sensitivity changes depending on the selected cutoff threshold for the value of the estimated function are shown in Figure 5.
As can be seen from the data in Figure 5, the values of balanced accuracy do not exceed 0.5. The minimum value of balanced accuracy is achieved at the threshold of the estimated function values equal to -8 (0.42), which corresponds to sensitivity values of 0.24 and specificity of 0.60. CONCLUSIONS Due to the severity of the SARS-CoV-2/COVID-19 coronavirus pandemic and the lack of sufficiently effective and safe therapeutic agents, the search for new pharmacological substances with anticoronavirus activity is extremely urgent [1]. D3Targets-2019-nCoV was the first freely available web resource that allows similarity assessment and molecular docking to identify compounds potentially suitable for COVID-19 therapy. This study evaluated the prediction quality using this platform for 94 substances of the test set containing 35 active compounds and 59 compounds, including compounds with established lack of activity against selected targets and compounds active against targets not represented in the CoViLigands database. We evaluated the reliability of the prediction using the parameter recommended by the authors, namely, the product of two-dimensional and three-dimensional similarity estimates (2D*3D). The prediction quality evaluation shows that the maximum balanced accuracy (0.57) is achieved at a similarity cutoff threshold of 31%. When calculating molecular docking's balanced accuracy, it is shown that its values do not exceed 0.5. Also, it was found that the maximum value of the balanced accuracy of the 2D similarity estimation method (0.59) is observed at the similarity cutoff threshold of 46%. For the 3D similarity estimation method, the maximum value of the balanced accuracy does not exceed 0.51. When analyzing the results obtained with the D3Similarity module, it was found that the 2D similarity score is 100% for identical structures. However, the 3D similarity score is lower. Considering that the structures of compounds uploaded for analysis and presented in the CoViLigands database undergo the same preprocessing, there is a question about the correctness of the methods chosen by the authors of the web service to analyze the similarity of three-dimensional structures. Based on the values of balanced accuracy for the entire range of predicted targets obtained on the test set formed by us and the established inconsistency in the results of structural similarity calculation, we can conclude the limited usefulness of the D3Target-2019-nCoV web service prediction. Thus, this resource requires a severe revision, including improving the used algorithms, particularly the methods of assessing 3D similarity. COMPLIANCE WITH ETHICAL STANDARDS This article does not contain any research involving humans or the use of animals as objects. FUNDING The work was supported by the Russian Foundation for Basic Research (project 20-04-60285). CONFLICT OF INTEREST The authors declare that they have no conflict of interest. SUPPLEMENTARY Supplementary materials are available at http://dx.doi.org/10.18097/BMCRM00140 REFERENCES
|