Гены «стахановцы» 18 хромосомы человека, отсутствующие белки и не охарактеризованные белки в ткани печени и клеточной линии hepg2
1Научно-исследовательский институт биомедицинской химии имени В. Н. Ореховича,
119121, Москва, ул. Погодинская, 10; e-mail: kseniadey@yandex.ru
2Институт молекулярной биологии им. В. А. Энгельгардта РАН, 119991, Москва, ул. Вавилова, 32
3Научно-исследовательский институт вакцин и сывороток им. И. И. Мечникова,
105064, Москва, Малый Казенный переулок, 5а
4Центр стратегического планирования и управления медико-биологическими рисками здоровью,
119121, Москва, ул. Погодинская, 10с1
6Кафедра судебной медицины, лечебного факультета, Российский научного-исследовательский
университет им. Н. И. Пирогова, 119034, Москва, пер. Хользунова, 7
7Сколковский институт науки и технологий, 121205,
Москва, территория Инновационного центра «Сколково», б-р Большой, 30с1
8Национальный исследовательский университет «Высшая школа экономики», 109028, Москва, б-р Покровский, 11
Ключевые слова: проект протеом человека; C-HPP; транскриптом; Oxford Nanopore Technologies; Illumina; РНК-секвенирование, протеотипические пептиды; отсутствующие белки
DOI:10.18097/BMCRM00144
Отсутствующие белки и функционально не охарактеризованные белки (в англоязычной литературе обозначенные как missing (MP) и functionally uncharacterized proteins (uPE1), соответственно) составляют менее 5% от общего числа белков, кодируемых генами 18 хромосомы человека. В течение полугода, начиная с января 2020 года, в версии NextProt выросло количество записей в наборах данных MP+uPE1. Подобные изменения обусловлены преимущественно достижениями протеомики на основе антител. В данной работе количественная ПЦР, технологии секвенирования Illumina HiSeq и Oxford Nanopore Technologies были применены для сравнительного анализа транскриптомного профиля образцов печени трех доноров мужского пола и клеточной линии HepG2. Анализ данных атласа экспрессии (Expression Atlas, EMBL-EBI) и полученных результатов по биологическим образцам с использованием ортогональных методов анализа транскриптома показал, что в клетках печени и HepG2 уровень экспрессии генов, кодирующих функционально не охарактеризованные белки (uPE1), находится на таком же низком уровне, как и в случае генов MP (в количестве менее 1 копии на клетку). Исключение составили несколько генов: HSBP1L1, TMEM241, C18orf21 и KLHL14. Согласно существенным расхождениям в ранее полученных полуколичественных данных по экспрессии генов и данным в открытых базах данных, изначально предполагалось, что экспрессия генов uPE1 может быть выше, чем генов MP. Подобное расхождение побудило обратиться к транскриптому 18 хромосомы человека, являющейся целевой для России в проекте «Протеом человека». Полученные результаты о наиболее экспрессируемых генах и дальнейший корреляционный анализ показал существование зависимости от метода экстракции мРНК и аналитической платформы.
Анализ экспрессии целевых генов 18 хромосомы с применением количественной ПЦР (qPCR) и методов высокопроизводительного профилирования транскриптома (Illumina HiSeq и ONT MinION) для одинаковых наборов образцов нормальной ткани печени и клеточной линии HepG2 выявил более 250 (92%) белок-кодирующих генов, детектируемых хотя бы одним методом. Экспрессия более чем 50% белок-кодирующих генов была детектирована всеми тремя методами. Корреляционный анализ профилей экспрессии генов показал, что результаты «группируются» в зависимости от типа биологического материала и экспериментальных методов, в частности от способа подготовки библиотеки (выделения кДНК, мРНК). Зависимость от выбора способа биоинформатической обработки была отмечена в значительно меньшей степени. Кроме того, комбинация методов секвенирования Illumina HiSeq и ONT MinION была использована для валидации протеотипических пептидов MP и uPE1 белков. Процедура была проделана для белка, связывающего фактор теплового шока HSBP1L1 (отсутствующий белок, недавно переведенный в категорию PE1) и не охарактеризованного C18orf21 (uPE1). Явление однонуклеотидного полиморфизма (SNP) приводило к утрате участка трипсинолизиса в HSBP1L1. Модифицированная версия HSBP1L1 была включена в базу данных с последующим поиском спектра MS/MS из набора данных Kulak, Geyer & Mann (2017), однако без существенных результатов идентификации. Таким образом, данные по масс спектрометрии для HSBP1L1 остаются не известными, хотя его существование на уровне белков было подтверждено.

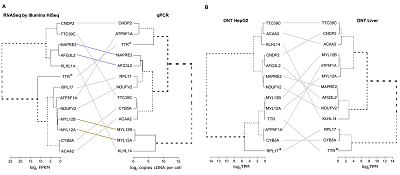
|
Рисунок 1.
Танглграмма наиболее высоко экспрессированных "стахановских" генов определённых с помощью (а) различных методов и одного образца (печень человека) и (б) с помощью одного и того же метода, примененного для разных биообразцов (печень и клеточная линия HepG2). Дендрограммы были получены с использованием разницы между оценками уровней экспрессии, измеренных методами qPCR, Illumina HiSeq и ONT, и масштабированы для удобства представления. * указывает на наиболее высоко экспрессированные гены. Дендрограммы были построены с использованием кластеризации Уорда с использование евклидовой метрики, расстояния рассчитывали для логарифмических величин (log2). Для построения танглграммы и расчета запутанности использовался пакет Dendextend [40].
|


|
Рисунок 2.
Корреляционная матрица между наборами данных по экспрессии генов, полученных для ткани печени и образцов клеточной линии HepG2 из разных источников и собранных в нашей предыдущей работе 2016 года (Hep13, Liv13) и этой работе (Hep20, Liv20). Индексы d1, d3 и d5 для Liv20 указывают на конкретных людей (доноров посмертных образцов печени). Наборы данных SRA: Hep13 - SRX395473 (2013) и SRX390071 (2014), Liv13 - SRX267708 (2013).
|
|
ЗАКРЫТЬ

|
Таблица 1.
Отобранные гены хромосомы 18 человека. Сначала два гена были аннотированы как кодирующие недостающие белки (MP), а четыре других гена были отнесены к категории функционально не охарактеризованных, но транслированных на белковый уровень с высокой степенью доказательности (uPE1). База знаний neXtProt была использована для получения информации о статусе гена, количестве аминокислотных остатков в последовательности изоформы 1 (самой длинной), одиночных аминокислотных полиморфизмах (SAPs) согласно наборам данных dbSNP и COSMIC, а также известных сплайс-формах (Iso1, Iso2, Iso3).
|
ФИНАНСИРОВАНИЕ
Работа выполнена при поддержке гранта Российского Научного Фонда #20-14-00328 с использованием оборудования центра «Геном» ЕИМБ РАН (http://www.eimb.ru/rus/ckp/ccu_genome_c.php).
ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ
К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi.org/10.18097/BMCRM00144) на сайте журнала.
ЛИТЕРАТУРА
- Paik, Y.K., Omenn, G.S., Hancock, W.S., Lane, L., Overall, C.M. (2017) Advances in the Chromosome-Centric Human Proteome Project: Looking to the Future. Expert Review of Proteomics, 1059–1071. DOI
- Omenn, G.S., Lane, L., Overall, C.M., Corrales, F.J., Schwenk, J.M., Paik, Y.K., Van Eyk, J.E., Liu, S., Pennington, S., Snyder, M.P., Baker, M.S., Deutsch, E.W. (2019) Progress on Identifying and Characterizing the Human Proteome: 2019 Metrics from the HUPO Human Proteome Project. Journal of Proteome Research, 4098–4107. DOI
- Gaudet, P., Argoud-Puy, G., Cusin, I., Duek, P., Evalet, O., Gateau, A., Gleizes, A., Pereira, M., Zahn-Zabal, M., Zwahlen, C., Bairoch, A., Lane, L. (2013) NeXtProt: Organizing Protein Knowledge in the Context of Human Proteome Projects. J. Proteome Res, 12 (1), 293–298. DOI
- Archakov, A., Aseev, A., Bykov, V., Grigoriev, A., Govorun, V., Ivanov, V., Khlunov, A., Lisitsa, A., Mazurenko, S., Makarov, A. A., Ponomarenko, E., Sagdeev, R., Skryabin, K. (2011) Gene-Centric View on the Human Proteome Project: The Example of the Russian Roadmap for Chromosome 18. Proteomics , 11 (10), 1853–1856. DOI
- Poverennaya, E.V., Ilgisonis, E.V., Ponomarenko, E.A., Kopylov, A.T., Zgoda, V.G., Radko, S.P., Lisitsa, A.V., Archakov, A.I. (2017) Why Are the Correlations between MRNA and Protein Levels so Low among the 275 Predicted Protein-Coding Genes on Human Chromosome 18? J. Proteome Res., 16 (12), 4311–4318. DOI
- Zgoda, V.G., Kopylov, A.T., Tikhonova, O.V., Moisa, A.A., Pyndyk, N.V., Farafonova, T.E., Novikova, S.E., Lisitsa, A.V., Ponomarenko, E.A., Poverennaya, E.V., Radko, S.P., Khmeleva, S.A., Kurbatov, L.K., Filimonov, A.D., Bogolyubova, N.A., Ilgisonis, E.V., Chernobrovkin, A.L., Ivanov, A.S., Medvedev, A.E., Mezentsev, Y.V., Moshkovskii, S.A., Naryzhny, S.N., Ilina, E.N., Kostrjukova, E.S., Alexeev, D.G., Tyakht, A.V., Govorun, V.M., Archakov, A.I. (2013) Chromosome 18 Transcriptome Profiling and Targeted Proteome Mapping in Depleted Plasma, Liver Tissue and HepG2 Cells. J. Proteome Res., 12 (1), 123–134. DOI
- Ponomarenko, E.A., Kopylov, A.T., Lisitsa, A.V., Radko, S.P., Kiseleva, Y.Y., Kurbatov, L.K., Ptitsyn, K.G., Tikhonova, O.V., Moisa, A.A., Novikova, S.E., Poverennay, E.V., Ilgisonis, E.V., Archakov, A.I. (2014) Chromosome 18 Transcriptoproteome of Liver Tissue and HepG2 Cells and Targeted Proteome Mapping in Depleted Plasma: Update 2013. J. Proteome Res., 13 (1), 183–190.
- Radko, S.P., Poverennaya, E.V., Kurbatov, L.K., Ponomarenko, E.A., Lisitsa, A.V., Archakov, A.I. (2019) The “Missing” Proteome: Undetected Proteins, Not-Translated Transcripts, and Untranscribed Genes. J. Proteome Res., 18 (12), 4273–4276. DOI
- Segura, V., Medina-Aunon, J. A., Guruceaga, E., Gharbi, S. I., Gonzälez-Tejedo, C., San Chez Del Pino, M.M., Canals, F., Fuentes, M., Ignacio Casal, J., Martínez-Bartolomé, S., Elortza, F., Mato, J. M., Arizmendi, J.M., Abian, J., Oliveira, E., Gil, C., Vivanco, F., Blanco, F., Albar, J.P., Corrales, F.J. (2013) Spanish Human Proteome Project: Dissection of Chromosome 16. J. Proteome Res., 12 (1), 112–122. DOI
- Yong-In, K., Jongan, L., Young-Jin, C., Jawon, S., Jisook, P., Soo-Youn, L., Je-Yoel, C. (2015) Proteogenomic Study Beyond Chromosome 9: New Insight Into Expressed Variant Proteome and Transcriptome in Human Lung Adenocarcinoma Tissues. J. Proteome Res., 14 (12), 5007–5016.
- Liu, S., Im, H., Bairoch, A., Cristofanilli, M., Chen, R., Deutsch, E. W., Dalton, S., Fenyo, D., Fanayan, S., Gates, C., Gaudet, P., Hincapie, M., Hanash, S., Kim, H., Jeong, S. K., Lundberg, E., Mias, G., Menon, R., Mu, Z., Nice, E., Paik, Y.K., Uhlen, M., Wells, L., Wu, S.L., Yan, F., Zhang, F., Zhang, Y., Snyder, M., Omenn, G.S., Beavis, R. C., Hancock, W.S. (2013) A Chromosome-Centric Human Proteome Project (C-HPP) to Characterize the Sets of Proteins Encoded in Chromosome 17. J Proteome Res, 45–57. DOI
- Chang, C., Li, L., Zhang, C., Wu, S., Guo, K., Zi, J., Chen, Z., Jiang, J., Ma, J., Yu, Q., Fan, F., Qin, P., Han, M., Su, N., Chen, T., Wang, K., Zhai, L., Zhang, T., Ying, W., Xu, Z., Zhang, Y., Liu, Y., Liu, X., Zhong, F., Shen, H., Wang, Q., Hou, G., Zhao, H., Li, G., Liu, S., Gu, W., Wang, G., Wang, T., Zhang, G., Qian, X., Li, N., He, Q. Y., Lin, L., Yang, P., Zhu, Y., He, F., Xu, P. (2014) Systematic Analyses of the Transcriptome, Translatome, and Proteome Provide a Global View and Potential Strategy for the C-HPP. J. Proteome Res., 13 (1), 38–49. DOI
- Shargunov, A.V., Krasnov, G.S., Ponomarenko, E.A., Lisitsa, A.V., Shurdov, M.A., Zverev, V.V., Archakov, A.I., Blinov, V.M. (2014) Tissue-Specific Alternative Splicing Analysis Reveals the Diversity of Chromosome 18 Transcriptome. J. Proteome Res., 13 (1), 173–182. DOI
- Krasnov, G.S., Dmitriev, A.A., Kudryavtseva, A.V., Shargunov, A.V., Karpov, D.S., Uroshlev, L.A., Melnikova, N.V., Blinov, V.M., Poverennaya, E.V., Archakov, A.I., Lisitsa, A.V., Ponomarenko, E.A. (2015) PPLine: An Automated Pipeline for SNP, SAP, and Splice Variant Detection in the Context of Proteogenomics. J. Proteome Res., 14 (9), 3729–3737. DOI
- Jeong, S.K., Lee, H.J., Na, K., Cho, J.Y., Lee, M.J., Kwon, J.Y., Kim, H., Park, Y.M., Yoo, J.S., Hancock, W. S., Paik, Y.K. (2013) GenomewidePDB, a Proteomic Database Exploring the Comprehensive Protein Parts List and Transcriptome Landscape in Human Chromosomes. J. Proteome Res., 12 (1), 106–111. DOI
- Poverennaya, E.V., Shargunov, A.V., Ponomarenko, E.A., Lisitsa, A.V. (2018) The Gene-Centric Content Management System and Its Application for Cognitive Proteomics. Proteomes, 6 (1). DOI
- Tyakht, A.V., Ilina, E.N., Alexeev, D.G., Ischenko, D.S., Gorbachev, A.Y., Semashko, T.A., Larin, A.K., Selezneva, O V., Kostryukova, E.S., Karalkin, P.A., Vakhrushev, I.V., Kurbatov, L.K., Archakov, A.I., Govorun, V.M. (2014) RNA-Seq Gene Expression Profiling of HepG2 Cells: The Influence of Experimental Factors and Comparison with Liver Tissue. BMC Genomics , 15 (1). DOI
- Chalmel, F., Rolland, A. D. Linking Transcriptomics and Proteomics in Spermatogenesis. Reproduction, 150 (5), R149–R157. DOI
- Fortelny, N., Overall, C. M., Pavlidis, P., Freue, G.V.C. (2017) Can We Predict Protein from MRNA Levels? Nature, 547, E19–E20. DOI
- Eraslan, B., Wang, D., Gusic, M., Prokisch, H., Hallström, B.M., Uhlén, M., Asplund, A., Pontén, F., Wieland, T., Hopf, T., Hahne, H., Kuster, B., Gagneur, J. (2019) Quantification and Discovery of Sequence Determinants of Protein‐per‐mRNA Amount in 29 Human Tissues. Mol. Syst. Biol., 15 (2). DOI
- De Sousa Abreu, R., Penalva, L.O., Marcotte, E.M., Vogel, C. (2009) Global Signatures of Protein and MRNA Expression Levels. Mol Biosyst, 1512–1526. DOI
- Ponomarenko, E.A., Kopylov, A.T., Lisitsa, A.V., Radko, S.P., Kiseleva, Y.Y., Kurbatov, L.K., Ptitsyn, K.G., Tikhonova, O.V., Moisa, A.A., Novikova, S.E., Poverennaya, E.V., Ilgisonis, E.V., Filimonov, A.D., Bogolubova, N.A., Averchuk, V.V., Karalkin, P.A., Vakhrushev, I.V., Yarygin, K.N., Moshkovskii, S.A., Zgoda, V.G., Sokolov, A.S., Mazur, A.M., Prokhortchouck, E.B., Skryabin, K.G., Ilina, E.N., Kostrjukova, E.S., Alexeev, D.G., Tyakht, A.V., Gorbachev, A.Y., Govorun, V.M., Archakov, A.I. (2014) Chromosome 18 Transcriptoproteome of Liver Tissue and HepG2 Cells and Targeted Proteome Mapping in Depleted Plasma: Update 2013. J. Proteome Res., 13 (1), 183–190. DOI
- Seki, M., Katsumata, E., Suzuki, A., Sereewattanawoot, S., Sakamoto, Y., Mizushima-Sugano, J., Sugano, S., Kohno, T., Frith C.,M., Tsuchihara, K., Suzuki, Y., Expand, A. (2019) Evaluation and Application of RNA-Seq by MinION. DNA Res ., 26 (1), 55–65.
- Poverennaya, E.V., Kopylov, A.T., Ponomarenko, E.A., Ilgisonis, E.V., Zgoda, V.G., Tikhonova, O.V., Novikova, S.E., Farafonova, T.E., Kiseleva, Y.Y., Radko, S.P., Vakhrushev, I.V., Yarygin, K.N., Moshkovskii, S.A., Kiseleva, O.I., Lisitsa, A.V., Sokolov, A.S., Mazur, A.M., Prokhortchouk, E.B., Skryabin, K.G., Kostrjukova, E.S., Tyakht, A.V., Gorbachev, A.Y., Ilina, E.N., Govorun, V.M., Archakov, A.I. (2016) State of the Art of Chromosome 18-Centric HPP in 2016: Transcriptome and Proteome Profiling of Liver Tissue and HepG2 Cells. J. Proteome Res., 15 (11), 4030–4038. DOI
- Riedel, G., Rüdrich, U., Fekete-Drimusz, N., Manns, M.P., Vondran, F.W.R., Bock, M. (2014) An Extended ΔCT-Method Facilitating Normalisation with Multiple Reference Genes Suited for Quantitative RT-PCR Analyses of Human Hepatocyte-like Cells. PLoS One, 9 (3). DOI
- Wilkening, S., Stahl, F., Bader, A. (2003) Comparison of Primary Human Hepatocytes and Hepatoma Cell Line HepG2 with Regard to Their Biotransformation Properties. Drug Metab. Dispos., 31 (8), 1035–1042. DOI
- Wick, R.R., Judd, L. M., Holt, K.E. (2019) Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol., 20 (1). DOI
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34 (18), 3094–3100. DOI
- Patro, R., Duggal, G., Love, M.I., Irizarry, R A., Kingsford, C. (2017) Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods, 14 (4), 417–419. DOI
- Kulak, N.A., Geyer, P.E., Mann, M. (2017) Loss-Less Nano-Fractionator for High Sensitivity, High Coverage Proteomics. Mol. Cell. Proteomics, 16 (4), 694–705. DOI
- Papatheodorou, I., Fonseca, N.A., Keays, M., Tang, Y. A., Barrera, E., Bazant, W., Burke, M., Füllgrabe, A., Fuentes, A.M.P., George, N., Huerta, L., Koskinen, S., Mohammed, S., Geniza, M., Preece, J., Jaiswal, P., Jarnuczak, A.F., Huber, W., Stegle, O., Vizcaino, J. A., Brazma, A., Petryszak, R. (2018) Expression Atlas: Gene and Protein Expression across Multiple Studies and Organisms. Nucleic Acids Res., 46 (D1), D246–D251. DOI
- Albert, R., Barabasi, A.L. (2002) Statistical mechanics of complex networks. Rev. Mod. Phys., 74(1), 47. DOI
- Poverennaya, E., Kiseleva, O., Ilgisonis, E., Novikova, S., Kopylov, A., Ivanov, Y., Kononikhin, A., Gorshkov, M., Kushlinskii, N., Archakov, Ponomarenko, E. (2020) Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins. Proteomes, 8 (2), 12. DOI
- Misawa, K., Kanazawa, T., Mochizuki, D., Imai, A., Mima, M., Yamada, S., Morita, K., Misawa, Y., Shinmura, K., Mineta, H. (2019) Genes Located on 18q23 Are Epigenetic Markers and Have Prognostic Significance for Patients with Head and Neck Cancer. Cancers, 11 (3). DOI
- Chen, K., He, Y., Liu, Y., Yang, X. (2019) Gene Signature Associated with Neuro-Endocrine Activity Predicting Prognosis of Pancreatic Carcinoma. Mol. Genet. Genomic Med., 7 (7). DOI
- Rodrigues, R.M., Heymans, A., De Boe, V., Sachinidis, A., Chaudhari, U., Govaere, O., Roskams, T., Vanhaecke, T., Rogiers, V., De Kock, J. (2016) Toxicogenomics-Based Prediction of Acetaminophen-Induced Liver Injury Using Human Hepatic Cell Systems. Toxicol. Lett., 240 (1), 50–59. DOI
- Deutsch, E.W., Lane, L., Overall, C. M., Bandeira, N., Baker, M. S., Pineau, C., Moritz, R.L., Corrales, F., Orchard, S., Van Eyk, J.E., Paik, Y.K., Weintraub, S.T., Vandenbrouck, Y., Omenn, G.S. (2019) Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J Proteome Res., 2019, 4108–4116. DOI
- Tran, J. C., Zamdborg, L., Ahlf, D. R., Lee, J.E., Catherman, A.D., Durbin, K. R., Tipton, J.D., Vellaichamy, A., Kellie, J.F., Li, M., Wu, C., Sweet, S.M.M., Early, B.P., Siuti, N., Leduc, R.D., Compton, P.D., Thomas, P.M., Kelleher, N.L. (2011) Mapping Intact Protein Isoforms in Discovery Mode Using Top-down Proteomics. Nature, 480 (7376), 254–258. DOI
- Righetti, P.G., Boschetti, E. (2008) The Proteominer and the Fortyniners: Searching for Gold Nuggets in the Proteomic Arena. Mass Spectrometry Reviews, 596–608. DOI
- Dendextend, G.T. (2015) Dendextend: An R Package for Visualizing, Adjusting and Comparing Trees of Hierarchical Clustering. Bioinformatics, 31 (22), 3718–3720. DOI
- Dong, H., Ge, X., Shen, Y., Chen, L., Kong, Y., Zhang, H., Man, X., Tang, L., Yuan, H., Wang, H., Zhao, G., Jin, W. (2009) Gene Expression Profile Analysis of Human Hepatocellular Carcinoma Using SAGE and LongSAGE. BMC Med. Genomics, 2. DOI
- 49 Shanmugam, A.K., Yocum, A.K., Nesvizhskii, A.I. (2014) Utility of RNA-Seq and GPMDB Protein Observation Frequency for Improving the Sensitivity of Protein Identification by Tandem MS. J. Proteome Res., 13 (9), 4113–4119. DOI
- Frith, M. C., Hamada, M., Horton, P. (2010) Parameters for Accurate Genome Alignment. BMC Bioinformatics, 11. DOI
- Li, H., Durbin, R. (2010) Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics, 26 (5), 589–595. DOI
- Van Delft, J., Gaj, S., Lienhard, M., Albrecht, M. W., Kirpiy, A., Brauers, K., Claessen, S., Lizarraga, D., Lehrach, H., Herwig, R., Kleinjans, J. (2012) Rna-Seq Provides New Insights in the Transcriptome Responses Induced by the Carcinogen Benzo[a]Pyrene. Toxicol. Sci., 130 (2), 427–439. DOI