Gut Microbiome and Drug Metabolism
Federal Research and Clinical Center of Physical-Chemical Medicine of FMBA,
1a Malaya Pirogovskaya str., Moscow, 119435 Russia; *e-mail: ilinaen@gmail.com
Key words: microbiota; metagenome; biotransformation; xenobiotics; microfluidics; computer modeling
DOI: 10.18097/BMCRM00146
The human physiology textbooks traditionally consider the intestine as a metabolically active organ, with its activity primarily associated with the production of numerous digestive enzymes. The development of molecular analysis technologies has significantly detailized this picture, primarily by decoding the metabolic potential of the intestinal microbiota. Data from numerous metagenomic studies indicate that the number of eukaryotic and bacterial cells in the human body is comparable - about 3.0×1013, while the number of genes in the intestinal metagenome is one hundred times greater than in the human genome. Obviously, the gut microbiota exhibits both direct and indirect effects on the metabolism of drugs and xenobiotics, that can affect their effectiveness and toxicity. Orally administrated xenobiotics have been found to be metabolized by intestinal microbial enzymes before being absorbed from the gastrointestinal tract into the blood flow. The metabolic reactions performed by the gut microbiota greatly differ from the metabolic reactions of the liver, providing modification of drugs by acetylation, deacetylation, decarboxylation, dehydroxylation, demethylation, dehalogenation, etc. Despite the metabolism of xenobiotics by microbial enzymes of the intestine is rather known, information about the specific microflora mediating each metabolic reaction is still limited, mainly by the lack of an adequate model of the intestinal microbial community to allow the accumulation of experimental data for the creation of computational models. Currently, studies of drug metabolism use microfluidic chips, reproducing functions of various organs and tissues, such as the liver, kidney, lungs and intestine, as in vitro models in the form of 2D and 3D cell cultures. Supplementation of such systems with the microbial community will allow to get as close as possible to in vitro modeling of complicated biological processes in the interests of pharmacological research and the accumulation of data for constructing computational models.
The original article is Russian. This version is a machine-generated translation with minimal terminological correction. If in doubt, use the original Russian version.
INTRODUCTION
The effectiveness of pharmacotherapy significantly varies from person to person. However, the recorded genetic differences between people are not sufficient to explain the entire spectrum of observed differences in responses to certain drugs [1].
Since most medications are taken orally, it is the gut microbiota that represents the most diverse and numerous microbial community, that has become the object of close attention of researchers. Increased interest in assessing the effect of microbiota on drug metabolism resulted in formation of a new direction of science - pharmacomicrobiomics, which was developed together with pharmacogenomics.
Data from numerous metagenomic studies indicate that the number of eukaryotic and bacterial cells in the human body is comparable – about 3.0x1013 [2]; at the same time, the number of genes in the human intestinal metagenome is about 530,000 versus 30,000 in the human genome [3]. According to the theory of functional redundancy [4], the intestinal ecosystem is formed by a "bacterial superspecies" with a very large genome, consisting of widely divergent microbial lines, whose genomes contain functionally similar sets of genes that can lead to a consistent single metabolic outcome.
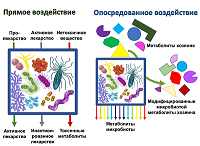
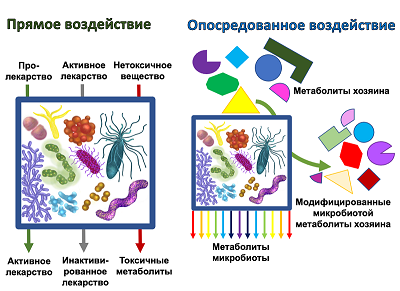
Considering the question of the effect on the metabolism of drugs, we can talk about various mechanisms which directly or indirectly involve gut microbiota. Direct exposure includes biotransformation, which leads to a change in the chemical structure of the drug compound, up to the degradation of the substance [5]. In some cases, biotransformation leads to the conversion of the prodrug into the active drug. However, in other cases, the drug is inactivated or converted to a toxic form. The variants of such interactions are shown schematically in Figure 1.
|
Figure 1.
Effect of microbiota on host metabolites and xenobiotics.
|
1. DIRECT BIOTRANSFORMATION OF MEDICINAL COMPOUNDS
Biotransformation of xenobiotics is one of the main functions of the intestinal microbiota. The intestine, both small and large, provides an environment with low redox potential and no or little available oxygen. Such anaerobic and regenerative conditions in the gut mediate the regenerative metabolism of incoming xenobiotics by resident intestinal bacteria.
Orally administered drugs can be metabolized by intestinal microbiota (for example, by reduction) and/or by enzymes that metabolize the host drugs (by oxidation), as a result of which they are bioactivated, eliminated or inactivated. On the other hand, drug use has been shown to be associated with the greatest inter-individual variability in the gut microbiota, and many non-antibiotic drugs have anticommensal activity, suggesting that some drugs cause changes the composition of the gut microbiota. In terms of the role of the gut microbiota in maintaining host health or developing disease, some prescription drugs (and other xenobiotics) may unintentionally affect host physiology through their effects on the composition/structure of the gut microbiota.
The metabolism of xenobiotics by the gut microbiota is known for decades and includes drugs of various chemical classes and therapeutic effects. Today, we can say that the gut microbiota as a single ecosystem contains enzymes that provide cascades of chemical reactions that lead to the biotransformation of a number of drugs (Table 1).

However, despite the accumulated evidence for xenobiotic biotransformation occurring under the influence of the intestinal microbiota, the biochemical and genetic bases of these processes are still largely unknown. This is partly due to the perceived difficulties of cultivating some gut bacteria and the extreme complexity of the gut microbiota community. For some drugs, the enzymes of certain gut bacteria responsible for their biotransformation have been identified and characterized, but for most compounds, only indirect evidence for involvement of intestinal bacteria-mediated regenerative metabolism is available. The physiological roles of even known intestinal bacterial enzymes are not fully defined.
At the same time, the contribution of the intestinal microbiota to the biotransformation of the drug sometimes dominates over the metabolic processes occurring under the action of host enzymes. Zimmermann and co-authors found that individual responses to the drug depended on the genetics of the human microbiota [7]. Using with different mutant microbiota they examined the metabolism of nucleoside drugs, used as antivirals and antidepressants, in mice. Then they modeled pharmacokinetics in different parts of the body and determined the contribution of the host and microorganisms. It was found that in some mice, up to 70% of drug biotransformation could be attributed to microbial metabolism [7].
Another consequence of the relationship between the genetic characteristics of the microbiota and the host's response to treatment is another approach to the search for therapy: the effect on the expression of certain genes in specific bacteria. Singh and co-authors have shown [8] that treatment of isolates of several multidrug-resistant bacteria with a double-acting immuno-antibiotic in the form of a prodrug produces an effect similar to conditional knockout of the target gene. The authors also noted that studied on a mouse this prodrug (4-hydroxy-3-methylbut2-enyl diphosphate reductase inhibitor, EC 1.17.1.2) stimulated the host immune response. Based on this, it can be assumed that a promising direction of drug development is to create a combination of the active substance itself and an auxiliary substance that modulates the expression of certain genes of specific bacteria, whose products contribute to reducing the effectiveness of the drug.
The human gut microbiome is known to metabolize many drugs [6]. Zimmermann and colleagues tested 76 human gut bacterial strains, representing 68 species from the main taxonomic groups of bacteria, to assess their ability to metabolize 271 drugs in vitro [9]. These drugs were selected to ensure the diversity of the group in terms of aspects such as molecular structure or effects on the body. It was found that 176 of the studied drugs underwent significant metabolic changes, caused by at least one bacterial strain, which led to a decrease in the activity level of the drug molecule. Each tested bacterial strain metabolized at least some of the drugs. At the same time, the number of drugs per strain varied from 11 to 95. Given that the authors tested a broadly representative group of drugs, the scale of these results is remarkable, as they increase the likelihood that most drugs are modified by the microbiota. After analyzing the biotransformation products of 176 preparations using mass spectrometry 868 unique molecules were identified. These data indicate that as a result of biotransformation by intestinal bacteria, more than one metabolite can be obtained from some drugs.
The ways of interaction are quite well studied for microbiota and various classes of antihypertensive drugs. Antihypertensive drugs, such as amlodipine and nifedipine, are metabolized by microbial enzymes in the intestine; this can affect the absorption of drugs and lead to changes in the pharmacological activity of the drug and possible failure of proper blood pressure control or unexpected side effects. This probably indicates the presence of additional mechanisms that alter the therapeutic effectiveness of antihypertensive drugs, especially with certain pathological conditions of the gastrointestinal tract or with the simultaneous use of substances such as antibiotics and probiotics, which can change the microbial composition of the intestine.
Most orally administered hydrophobic xenobiotics are converted to hydrophilic products by oxidation and conjugation, facilitating their elimination and detoxification. These processes occur mainly in the liver. However, it has been found that xenobiotics administered orally can also be metabolized by intestinal microbial enzymes before being absorbed from the gastrointestinal tract into the blood. The metabolic reactions performed by the gut microbiota differ significantly from the metabolic reactions of the liver. Microbiota of the gastrointestinal tract metabolizes drugs and phytochemicals mainly to hydrophobic compounds, that affect the absorption of drugs and can change their pharmacological effects. This involves the main microbial enzymes, such as β-glucosidases, β-glucuronidases, sulfatases, azoreductases, nitroreductases, and nitrate reductases. Typical examples of compounds and drugs metabolized by the gut microbiota include simvastatin, prontosil, irinotecan, glycyrrhizin, amygdalin, baykalein, ginsenosides, and genistein. Some drugs, such as like acetaminophen, amlodipine, chloramphenicol, digoxin, lovastatin, and sulfasalazine are also affected by intestinal microbial metabolism (Table 1).
Although metabolism of xenobiotics by gut microbial enzymes is known to some extent, information about the specific microflora whose enzymes catalyze each metabolic reaction is still limited. The microbiota has a complex effect on the metabolism and transport of orally administered medicinal compounds and on the metabolic activity of the intestinal mucosa. Orally administered drugs, especially those that are not completely absorbed from the small intestine or candidates for enterohepatic circulation, inevitably contact with the microbiota of the distal part of the intestine.
The gut microbiota has a diverse and extensive biotransformation capacity for the metabolism of xenobiotic compounds. Conditions that affect the microbial profile of the gut, such as animal sterilization or antibiotic treatment, can lead to changes in the pharmacokinetics of xenobiotic compounds, including phytochemicals, dietary compounds, and many medications. Although a very limited number of studies have directly investigated the effect of the gut microbiota on pharmacokinetics of antihypertensive drugs, the microbiota is known to affect the bioavailability of antihypertensive drugs in various ways, including bacterial metabolism, modulation of metabolic enzymes, and intestinal transport. This involvement of the gut microbiota may indicate additional mechanisms that may alter the therapeutic efficacy of antihypertensive drugs, especially in certain pathologic conditions of the gastrointestinal tract or with the simultaneous use of substances such as antibiotics and probiotics that may alter the microbial composition of the gut. Under these possibilities, future studies will focus more on the potential metabolism of antihypertensive drugs by the gut microbiota [10].
Advances in metagenomics, metabolomics, and bioinformatics, as well as the collection of thousands of gut bacteria and improved cultivation methods provided huge resources for identification of specific gut bacteria and their enzymes responsible for drug biotransformation. However, for most of the reductive metabolites described in the literature, gut bacteria and their enzymes mediating these reactions still remain unknown. Identifying and characterizing these gut bacteria and their enzymes will not only provide a basis for the integration of the intestinal microbiota into the optimization of the results of drug therapy in the host, but will also provide an opportunity to determine their physiological role in bacteria and in the intestinal community [11].
2. INDIRECT INFLUENCE OF THE MICROBIOTA FOR DRUG THERAPY
The influence of microbiota for antitumor drugs is of particular interest. For example, colorectal cancer clarified some important nuances: first, bacteria play unequal role in tumor development and interact with each other (the concept of "driver-passenger", with some bacteria contribute to the development of tumors, while others settle on them how comfortable dwelling place), and secondly, the composition of the local community and their substrate change with the development of tumor, because microorganisms modify their local biotope; third, the bacteria in the tumor are unevenly distributed [12], which can have a local effect on metabolic processes. At the same time, the results of local influences can be combined due to the fact that these local microsystems are not isolated from each other. In addition, the combination of metabolites of several bacteria can lead to different variants of biotransformation compared to biotransformation by metabolites of the same bacteria separately. To study the heterogeneity of the distribution of individual bacterial species, for example, HiPR-FISH technology has been used to identify and map the spatial distribution of microorganisms in a complex community in high resolution [13].
It was previously observed [14] that in vitro cultivation of two types of human tumor cells together with fibroblasts led to unexpected survival of tumor cells after exposure to the chemotherapeutic drug gemcitabine. Geller and her colleagues conducted research on this phenomenon. Using DNA sequence analysis, they found that the fibroblast sample was infected with the Mycoplasma hyorhinis [15]. Other examples have been reported where bacteria modulated the effect of antitumor therapy. For example, in patients with colorectal cancer the colon often has a large number of Fusobacterium nucleatum associated with tumors, and this is associated with a worse clinical prognosis [16]. In colorectal cancer cells, this bacterium can activate pathways that give the tumor resistance to the drugs fluorouracil and oxaliplatin [17].
It is worth noting that the intestinal microbiota can influence both the response of the host body, and the drug and its toxicity, modulating the molecular pathways of the host responsible for the metabolism and transport of drugs. This effect can be carried out by influencing the expression of host genes involved in drug transport and metabolism, as well as by interfering with the host's enzymatic activity and modulating immune responses [18, 19]. Such effects may be mediated by microbial metabolites or modification of host metabolites, in particular including their direct biotransformation after isolation them with bile from the liver. The isolation of the drug metabolites with bile provides an opportunity for their repeated biotransformation by beta-glucuronidase enzymes, which can reactivate the drug in the intestine, causing its increased toxicity. An example of this phenomenon is irinotecan (CPT-11), a widely used intravenous prodrug for the treatment of colon cancer. After the introduction of irinotecan, a complex series of biotransformation reactions occurs. Thus, as a result of irinotecan hydrolysis of carboxylesterase forms its active metabolite SN-38, which exhibits toxicity to intestinal epithelial cells and is believed by researchers to exacerbate diarrhea observed in almost 80% of patients [20]. A similar mechanism contributes to the occurrence of side effects when taking nonsteroidal anti-inflammatory drugs, including diclofenac, indomethacin, and ketoprofen [21].
A comparison of germ-free and normal mice showed that the microbiota affects the expression of a number of host genes involved in the metabolism and transport of drugs [18]. This effect on the expression of host genes by the gut microbiota can be local [22] (in intestinal tissues) or indirect, which includes damage to the liver as an organ involved in drug metabolism [23].
A comparison of germ-free and normal mice showed that the gut microbiota not only changed the spectrum of endogenous metabolites (10% of total metabolites they differ quantitatively by at least 50%), but also contributed to the unique microbial compounds circulating in the circulatory system [24, 25]. Some of these microbial metabolites were processed by the host in the same way as xenobiotics [24]. This overlap in the host's responses to drugs and microbial metabolites may have consequences during drug therapy; for example, it may lead to increased toxicity or half-life of xenobiotics as a result of competition between the drug and microbial metabolites for the same host enzymes that are involved in the detoxification or elimination of drugs.
Drugs in the body pass through a complex path associated with redistribution and metabolic transformations, and eventually - excrete. Before entering the circulatory system and reaching the target tissue or organ, orally administered substances are metabolized in the intestine and liver, with reducing the possible systemic concentration of drugs and, as a result, their effectiveness. At the same time, another indicator, the bioavailability of the drug, depends, among other things, on the activity of enzymes in the intestine and liver [26]. The activity of these enzymes can vary up to ten-fold between individuals [27], which highlights significant inter-individual differences in metabolism by intestinal microorganisms that may contribute to variations in the effectiveness of the drug [28].
When evaluating the processes of interaction between the microbiome and drugs in the body, it is necessary to keep in mind the distinct differences in pharmacological groups. The effect of the gut microbiota on antitumor therapy may be mediated through mechanisms such as general immunomodulation, an increase in the number of cells, specifically reacting to antigens of both microbial and tumor origin, metabolism, degradation (disposal) of medicines. Currently, the gut microbiota is considered as an additional, but important target object for studying the effectiveness of antitumor therapy and reducing its toxicity, as well as a predictor of the success of immunotherapy [29].
The mutual influence of the applied immunotherapy and the intestinal microbiota can lead to the emergence of secondary resistance to antitumor therapy, or vice versa - an increase in antitumor effect. The complex relationships between the host immune system, the course of the tumor process in accordance with the biology of a particular type of tumor, the commensal and tumor microbiota, and therapeutic measures are based on a variety of molecular mechanisms that attract the attention of researchers.
In addition, the tools and methods used to characterize the microbiome are constantly evolving to provide more data at a lower cost. New approaches in the immunotherapy of malignant neoplasms have indicated the presence of a link between the gut microbiome and the success of therapeutic measures. The identification of the exact molecular mechanisms by which the microbiota mediates the response of a particular type of tumor to immuno-chemotherapy will make it possible to widely use the means of microbiome modulation in order to increase the therapeutic effect and reduce the toxicity of antitumor drugs.
In particular, anti-PD1 immunotherapy aimed at programmed cell death protein 1 (PD1) has shown great potential in the treatment of malignant neoplasms, but its success depends on many factors, one of which is the composition of the intestinal microbiota. A study of the gut microbiota in patients with melanoma has shown that there is a relationship between the gut microbiome and the response to PD-1 immunotherapy in these patients [29-31]. Patients with a favorable gut microbiome may have enhanced systemic and antitumor immune responses mediated by increased antigen presentation and improved function of effector T cells in the tumor microenvironment.
In a recent study performed on a Russian cohort of patients, it was possible to detect marker microorganisms associated with the response to anti-PD1 therapy, including those similar to previously detected [31].
In general, a positive effect on the success of anti-PD1 therapy associated with “more healthy" composition of the microbiome, which contributes to better digestion and assimilation of nutrients from food, in particular - of plant origin. The therapeutic potential of modulation of the intestinal microbiome in patients receiving immunotherapy with checkpoint blockade may be important for the success of treatment and prognosis of the course of the tumor process, which will require rapid assessment of the microbiome in cancer patients during therapy.
Considering the effect of the microbiota on metabolism, it is necessary to take into account that xenobiotics themselves can affect the intestinal microbiota. It is known at least 30 drugs that directly affect the activity of the intestinal microbiome [32]. Such drugs can induce changes in gene expression in representatives of several taxonomic groups of the microbiome [33], which, in turn, can lead to a decrease in the production of enzymes required for biotransformation. Apart from the well-known action of antibiotics on microbiota, there is strong evidence that the impact of xenobiotics such as heavy metals, pesticides, nanoparticles, polycyclic aromatic hydrocarbons, dioxins, furans, polychlorinated biphenyls and "low-calorie" artificial sweeteners affect the microbiome and this probably contributes to the development of metabolic, malignant, inflammatory, or immune diseases [34]. Moreover, such xenobiotics can get directly from the external environment (for example, when breathing), and due to poor quality cleaning of raw materials that have been used for the production of food or medicines consumed by humans.
It is known that the structure of the microbiome is closely related to diet [35], age [36] and the genetics of the host itself [37]. In particular, correlations were found between the presence or absence of certain bacterial taxons and diet, as well as associations mediated by the gut microbiota between diet and circulating metabolites in women with low bone mineral density [38].
Here is another example of the relationship between diet, microbiota, and circulating metabolites. The cardiac glycoside digoxin is inactivated by the "cardiac glycoside reductase" of Eggerthella lenta. This undesirable inactivation can be reduced by increasing the amount of arginine in food, which indicates the effect of dietary interventions on the interaction of the drug with the microbiota [39]. Only E. lenta strains, carrying "cardiac glycoside reductase", carry out this biotransformation. The abundance of "cardiac glycoside reductase" in stool samples can predict the inactivation of digoxin and, as a result, a decrease in the activity of drugs [39]. This example clearly demonstrates that genomic and transcriptomic analyses of the gut microbiota can be useful for prediction of drug reactions. All these data indicate the need to implement an integrated approach.
3. MODELING CAPABILITIES METABOLISM OF MEDICINAL COMPOUNDS
Taking into account the wide range of drugs that are metabolized by the gut microbiota, it is extremely important to study the maximally possible library of drugs in order to develop a personalized approach in medicine. This is especially important due to the fact that the metabolic pathways of drugs often coincide. This allows to build computational algorithms enabling to predict the modifications of drugs that will be produced by the microbiota of a particular patient. In [40], the authors built a similar computing system called microbeFDT. First, they evaluated the degree of chemical similarities between 10,822 different substances, including components of food, drugs, and endogenous compounds. The similarity of the compounds was calculated on the basis of calculation of common substructures. The authors then used data from the Integrative Human project Microbiome Project (iHMP) [41] to determine the known enzymes involved in the metabolism of the chemical compounds considered, as well as the bacteria containing the genes encoding these enzymes in their genomes.
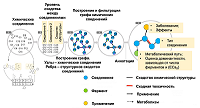
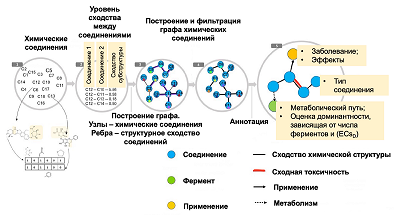
For the subsequent analysis, a graph was constructed (Fig. 2) containing the following information: 1) similarity of chemical structures of medicinal products, 2) the side effects described for them, 3) the enzymes involved in the metabolic transformations of these substances. This made it possible to predict the metabolism of unknown compounds in the case when a similar substance was known with known metabolism performed by microbes.
|
Figure 2.
Schematic representation of the chemical compounds graph structure used to predict drug metabolism in microbeFDT software. The main stages of graph construction are shown: 1) analysis of chemical structures of compounds and identification of common substructures; 2) calculation of the level of similarity of compounds based on the presence of common substructures: 3) construction of a graph in which the nodes correspond to chemical compounds, and the edges contain information about the degree of similarity between them; 4) filtering the graph, removing substances for which no structurally related compounds were found; 5) adding nodes to the graph containing information about medicinal and other effects of chemical compounds and enzymes that can metabolize these compounds. Adapted from [40].
|
Applicability of the approach was demonstrated by analysis and experimental testing of the microbial metabolism of the antitumor drug altretamine, used in the treatment of ovarian cancer. Approximately half of the patients treated with oral administration of altretamine had various toxic manifestations from the gastrointestinal tract. The microbeFDT system was used to detect a structurally similar drug to altretamine, melamine, exhibiting known microbiota-mediated toxicity. Based on the constructed graph, N-demethylase putative enzyme responsible for demethylation of this compound was found. Incubation of altretamine with fecal suspension and LC-MS analysis confirmed the presence of this modification. The influence on the microbial community (destruction of living microorganisms) significantly reduced the level of observed modification.
The lack of realistic mechanistic models significantly hampers understanding of the heterogeneity of human reactions to pharmacological agents. Although pharmacokinetic models describe in details the distribution in the body and metabolism of drugs, they do not take into account individual variations of the microbiome in addition to genetic variations.
Recent advances in the development of microfluidic systems may provide new approaches to the analysis of medicinal compounds in vitro, including their screening, active testing, and the study of metabolism [42]. To date, the functions of various organs and tissues, such as the liver [42], kidneys [43], lungs, and intestine, are reproduced in the form of in vitro models, 2D and 3D (spheroids, organoids) cell cultures. Co-cultivation of several types of functional cells in “organ-on-a-chip” microfluidic systems can be considered as a new variation of screening platforms ADME (absorption, metabolism, distribution and excretion); using this approach simultaneous and complex analysis of cells, drugs and their metabolites becomes quite possible. To predict complex organ interactions, a "body-on-a-chip" system has been proposed; it integrates functions of several organs on a single microfluidic device [44, 45]. This approach looks particularly promising in relation to the study of oncological diseases [46-48].
Maximum approximation of the biotransformation model to adapt medicinal compounds to in vivo conditions is impossible without introducing microbiome components into such devices. For many years, researchers have been developing systems for in vitro gut microbiota modeling by using various types of bioreactors, as well as using cellular substrates that are designed to mimic the cellular diversity present in the human gut, the flow of metabolites, and peristaltic movements.
Among the bioreactors maintaining continuous cultures, there are chemostats, auxostats and retentostats. They all consist of tanks, sensors, a system for entering the nutrient medium and removing waste products to maintain stable conditions, but the methods of creating stability are different. In chemostats, the specific growth rate is regulated by controlling the rate of adding fresh media and removing waste products and a part of the cells. A stable population density is maintained in auxostats [49]. In retentostats that represent modified chemostats, the waste water outflow occurs, while the biomass is retained with the help of special filters. Stability in the retentostats is maintained by the limited food resource. Therefore, the growth rate of microorganisms in them is close to zero [50]. Also mechano-chemostats are described; these microfluidic devices may be used to study the effect of mechanical stress on the growth of microorganisms [51]. A recently described device "Omnistat" (Omnistat) can work as a chemostat, as an aucostat, and as a retentostat, depending on the settings (temperature, pH, oxygenation, fresh water flow environment, etc.). This system provides the possibility of flows between vessels operating in different modes and supporting different cultivation conditions [52].
It is obvious that adequate in vitro modeling of the gut microbiome requires creation of models using cell biology technologies, among which intestinal organoids can be considered to be the most promising ones. Organoids are created from induced stem cells that under certain culture conditions proliferate and form spheres. In these areas, the intestinal epithelium, mesenchyme, and structures similar to the intestinal lumen are present [53]. Organoids as a model are used for a wide range of studies: from the interaction of intestinal tissues with the microbiome [54] to the study viral [55] and parasitic infections.
A more advanced technology, similar to organoids, is creation of an organ model on an artificial frame-matrix [56]. Such models can be created from various cell lines, including their combinations, and also allow the creation of hybrid organs [57]. At its core, this technology is a universal solution for any research tasks; it is limited only by the complexity of the framework-matrix. As already mentioned above, all kinds of microfluidic devices that implement “organ-on-a-chip” systems now are actively developed [58]. Currently, there is a number of such devices [59], however, the HuMiX microfluidic device attracts special attention [60, 61]. The device is designed to study the interaction of the microbiota and the human gastrointestinal tract and consists of three compartments separated by semipermeable membranes, which allow both eukaryotic cells and microbial strains to be cultured simultaneously. Other devices allow the co-cultivation of complex communities of anaerobic and aerobic commensal bacteria and intestinal epithelium [62].
Today, we are still far from creating reliable solutions of microfluidic “body-on-a-chip” devices that implement heterologous ecosystems similar to the gut microbiome. However, the creation of such devices, starting with in vitro models of the intestine, is extremely important for the transition to high-performance testing of the effect of intestinal microorganisms on a wide range of xenobiotics (and vice versa), including detailed analysis of structural and functional properties and the construction of predictive computational models.
CONCLUSIONS
Currently, the problem of the influence of the microbiota on drug metabolism of drugs remains insufficiently studied, but there are prerequisites for a breakthrough in this area. Based on the described above, we can conclude that the study of the influence of the microbiota on the drug effectiveness requires a comprehensive approach. The development of complex controlled models will allow us to clarify in detail the mechanisms of action of medicines, which, in turn, will make it possible to make a more balanced decision on the appointment of a particular drug tools. The data obtained in the course of such studies will allow not only to improve existing therapeutic methods by identifying new factors that affect the effectiveness of a particular drug, but also to develop fundamentally new approaches to treatment.
Complex microfluidic systems based on the "body-on-a-chip" type will allow conducting studies with a high level of control of conditions, with a low level of simplification of the system relative to a living organism, and in high resolution.
FUNDING
The work was carried out with financial support Ministry of Science and Higher Education (Agreement RFMEFI60419X0215).
COMPLIANCE WITH ETHICAL STANDARDS
This article does not contain any research involving people or using animals as objects.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
REFERENCES
- Nebert, D.W., Zhang, G., Vesell, E.S. (2008). From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metabolism Reviews, 40(2), 187–224. DOI
- Sender, R., Fuchs, S., & Milo, R. (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biology, 14(8), e1002533. DOI
- Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., Nielsen, T., Pons, N., Levenez, F., Yamada, T., Mende, D. R., Li, J., Xu, J., Li, S., Li, D., Cao, J., Wang, B., Liang, H., Zheng, H., Xie, Y., Tap, J., Lepage, P., Bertalan, M., Batto, J.M., Hansen, T., Le Paslie,r D., Linneberg, A., Nielsen, H.B., Pelletier, E., Renault, P., Sicheritz-Ponten, T., Turner, K., Zhu, H., Yu, C., Li, S., Jian, M., Zhou, Y., Li, Y., Zhang, X., Li S., Qin, N., Yang, H., Wang, J., Brunak, S., Doré J., Guarner, F., Kristiansen, K., Pedersen, O., Parkhill, J., Weissenbach, J., MetaHIT Consortium, Bork, P., Ehrlich, S.D., Wang, J. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 464(7285), 59–65. DOI
- Moya, A., & Ferrer, M. (2016). Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends in Microbiology, 24(5), 402–413. DOI
- Spanogiannopoulos, P., Bess, E. N., Carmody, R. N., & Turnbaugh, P. J. (2016). The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nature Reviews. Microbiology, 14(5), 273–287. DOI
- Haiser, H. J., & Turnbaugh, P. J. (2013). Developing a metagenomic view of xenobiotic metabolism. Pharmacological Research, 69(1), 21–31. DOI
- Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R., & Goodman, A. L. (2019). Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science (New York, N.Y.), 363(6427), eaat9931. DOI
- Singh, K. S., Sharma, R., Reddy, P., Vonteddu, P., Good, M., Sundarrajan, A., Choi, H., Muthumani, K., Kossenkov, A., Goldman, A. R., Tang, H. Y., Totrov, M., Cassel, J., Murphy, M. E., Somasundaram, R., Herlyn, M., Salvino, J. M., & Dotiwala, F. (2021). IspH inhibitors kill Gram-negative bacteria and mobilize immune clearance. Nature, 589(7843), 597–602. DOI
- Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R., Goodman, A.L. (2019). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 570(7762), 462-467. DOI
- Choi, M. S., Yu, J. S., Yoo, H. H., & Kim, D. H. (2018). The role of gut microbiota in the pharmacokinetics of antihypertensive drugs. Pharmacological Research, 130, 164–171. DOI
- Guo, Y., Lee, H., & Jeong, H. (2020). Gut microbiota in reductive drug metabolism. Progress in Molecular Biology and Translational Science, 171, 61–93. DOI
- Wang, Y., Zhang, C., Hou, S., Wu, X., Liu, J., & Wan, X. (2020). Analyses of Potential Driver and Passenger Bacteria in Human Colorectal Cancer. Cancer Management and Research, 12, 11553–11561. DOI
- Shi, H., Shi, Q., Grodner, B., Lenz, J. S., Zipfel, W. R., Brito, I. L., & De Vlaminck, I. (2020). Highly multiplexed spatial mapping of microbial communities. Nature, 588(7839), 676–681. DOI
- Straussman, R., Morikawa, T., Shee, K., Barzily-Rokni, M., Qian, Z. R., Du, J., Davis, A., Mongare, M. M., Gould, J., Frederick, D. T., Cooper, Z. A., Chapman, P. B., Solit, D. B., Ribas, A., Lo, R. S., Flaherty, K. T., Ogino, S., Wargo, J. A., & Golub, T. R. (2012). Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature, 487(7408), 500–504. DOI
- Geller, L. T., Barzily-Rokni, M., Danino, T., Jonas, O. H., Shental, N., Nejman, D., Gavert, N., Zwang, Y., Cooper, Z. A., Shee, K., Thaiss, C. A., Reuben, A., Livny, J., Avraham, R., Frederick, D. T., Ligorio, M., Chatman, K., Johnston, S. E., Mosher, C. M., Brandis, A., Fuks, G., Gurbatri, C., Gopalakrishnan, V., Kim, M., Hurd, M.W., Katz, M., Fleming, J., Maitra, A., Smith, D.A., Skalak, M., Bu, J., Michaud, M., Trauger, S.A., Barshack, I., Golan, T., Sandbank, J., Flaherty, K.T., Mandinova, A., Garrett, W.S., Thayer, S.P., Ferrone, C.R., Huttenhower, C., Bhatia, S.N., Gevers, D., Wargo, J.A., Golub, T.R., Straussman, R. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science, 357(6356), 1156–1160. DOI
- Mima, K., Nishihara, R., Qian, Z. R., Cao, Y., Sukawa, Y., Nowak, J. A., Yang, J., Dou, R., Masugi, Y., Song, M., Kostic, A. D., Giannakis, M., Bullman, S., Milner, D. A., Baba, H., Giovannucci, E. L., Garraway, L. A., Freeman, G. J., Dranoff, G., Garrett, W. S., Huttenhower, C., Meyerson, M., Meyerhardt, J.A., Chan, A.T., Fuchs, C.S., Ogino, S. (2016). Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut, 65(12), 1973–1980. DOI
- Yu, T., Guo, F., Yu, Y., Sun, T., Ma, D., Han, J., Qian, Y., Kryczek, I., Sun, D., Nagarsheth, N., Chen, Y., Chen, H., Hong, J., Zou, W., Fang, J. Y. (2017). Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell, 170(3), 411-413. DOI
- Björkholm, B., Bok, C. M., Lundin, A., Rafter, J., Hibberd, M. L., & Pettersson, S. (2009). Intestinal microbiota regulate xenobiotic metabolism in the liver. PloS One, 4(9), e6958. DOI
- Clayton, T. A., Baker, D., Lindon, J. C., Everett, J. R., & Nicholson, J. K. (2009). Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proceedings of the National Academy of Sciences of the United States of America, 106(34), 14728–14733. DOI
- Stein, A., Voigt, W., & Jordan, K. (2010). Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Therapeutic Advances in Medical Oncology, 2(1), 51–63. DOI
- Higuchi, K., Umegaki, E., Watanabe, T., Yoda, Y., Morita, E., Murano, M., Tokioka, S., & Arakawa, T. (2009). Present status and strategy of NSAIDs-induced small bowel injury. Journal of Gastroenterology, 44(9), 879–888. DOI
- Hooper, L. V., Wong, M. H., Thelin, A., Hansson, L., Falk, P. G., & Gordon, J. I. (2001). Molecular analysis of commensal host-microbial relationships in the intestine. Science, 291(5505), 881–884. DOI
- Claus, S. P., Ellero, S. L., Berger, B., Krause, L., Bruttin, A., Molina, J., Paris, A., Want, E. J., de Waziers, I., Cloarec, O., Richards, S. E., Wang, Y., Dumas, M. E., Ross, A., Rezzi, S., Kochhar, S., Van Bladeren, P., Lindon, J. C., Holmes, E., & Nicholson, J. K. (2011). Colonization-induced host-gut microbial metabolic interaction. mBio, 2(2), e00271-10. DOI
- Wikoff, W. R., Anfora, A. T., Liu, J., Schultz, P. G., Lesley, S. A., Peters, E. C., & Siuzdak, G. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the National Academy of Sciences of the United States of America, 106(10), 3698–3703. DOI
- Claus, S. P., Tsang, T. M., Wang, Y., Cloarec, O., Skordi, E., Martin, F. P., Rezzi, S., Ross, A., Kochhar, S., Holmes, E., & Nicholson, J. K. (2008). Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Molecular systems biology, 4, 219. DOI
- Pond, S. M., & Tozer, T. N. (1984). First-pass elimination. Basic concepts and clinical consequences. Clinical Pharmacokinetics, 9(1), 1–25. DOI
- Deloménie, C., Fouix, S., Longuemaux, S., Brahimi, N., Bizet, C., Picard, B., Denamur, E., & Dupret, J. M. (2001). Identification and functional characterization of arylamine N-acetyltransferases in eubacteria: evidence for highly selective acetylation of 5-aminosalicylic acid. Journal of Bacteriology, 183(11), 3417–3427. DOI
- Sousa, T., Yadav, V., Zann, V., Borde, A., Abrahamsson, B., & Basit, A. W. (2014). On the colonic bacterial metabolism of azo-bonded prodrugsof 5-aminosalicylic acid. Journal of Pharmaceutical Sciences, 103(10), 3171–3175. DOI
- Olekhnovich, E. I., Manolov, A. I., Pavlenko, A. V., Konanov, D. N., Fedorov, D. E., Tikhonova, P. O., Glushchenko, O. E., Ilina, E. N. (2020). Intestinal microbiom modulates the response to antitumor immunotherapy. Biomeditsinskaya Khimiya, 66(1), 54-63. DOI
- Hamid, O., Robert, C., Daud, A., Hodi, F. S., Hwu, W. J., Kefford, R., Wolchok, J. D., Hersey, P., Joseph, R. W., Weber, J. S., Dronca, R., Gangadhar, T. C., Patnaik, A., Zarour, H., Joshua, A. M., Gergich, K., Elassaiss-Schaap, J., Algazi, A., Mateus, C., Boasberg, P., … Ribas, A. (2013). Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England Journal of Medicine, 369(2), 134–144. DOI
- Fedorov, D. E., Olekhnovich, E. I., Pavlenko, A. V., Klimina, K. M., Pokataev, I. A., Manolov, A. I., Konanov, D. N., Veselovsky, V. A., Ilina, E. N. (2020). Intestinal microbiome as a predictor of the anti-PD-1 therapy success: metagenomic data analysis. Biomeditsinskaya Khimiya, 66(6), 502-507. DOI
- Klaassen, C. D., & Cui, J. Y. (2015). Review: Mechanisms of How the Intestinal Microbiota Alters the Effects of Drugs and Bile Acids. Drug Metabolism and Disposition, 43(10), 1505–1521. DOI
- Maurice, C. F., Haiser, H. J., & Turnbaugh, P. J. (2013). Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell, 152(1-2), 39–50. DOI
- Tsiaoussis, J., Antoniou, M. N., Koliarakis, I., Mesnage, R., Vardavas, C. I., Izotov, B. N., Psaroulaki, A., & Tsatsakis, A. (2019). Effects of single and combined toxic exposures on the gut microbiome: Current knowledge and future directions. Toxicology Letters, 312, 72–97. DOI
- David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., Ling, A. V., Devlin, A. S., Varma, Y., Fischbach, M. A., Biddinger, S. B., Dutton, R. J., & Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505(7484), 559–563. DOI
- Frommknecht H. (1988). Der Arzt--Partner der Privaten Krankenversicherung [The physician--the partner of private health insurance]. Versicherungsmedizin, 40(2), 33–34.
- Usenbaev A. (1971). Blood indices and their changes following blood giving by donors in different geographic localities. Sovetskoe zdravookhranenie Kirgizii, 1, 3–7.
- Palacios-González, B., Ramírez-Salazar, E. G., Rivera-Paredez, B., Quiterio, M., Flores, Y. N., Macias-Kauffer, L., Moran-Ramos, S., Denova-Gutiérrez, E., Ibarra-González, I., Vela-Amieva, M., Canizales-Quinteros, S., Salmerón, J., Velázquez-Cruz, R. (2020). A multi-omic analysis for low bone mineral density in postmenopausal women suggests a relationship between diet, metabolites, and microbiota. Microorganisms, 8(11), 1630. DOI
- Haiser, H. J., Gootenberg, D. B., Chatman, K., Sirasani, G., Balskus, E. P., & Turnbaugh, P. J. (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science, 341(6143), 295–298. DOI
- Guthrie, L., Wolfson, S., & Kelly, L. (2019). The human gut chemical landscape predicts microbe-mediated biotransformation of foods and drugs. eLife, 8, e42866. DOI
- Integrative HMP (iHMP) Research Network Consortium (2014). The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host and Microbe, 16(3), 276–289. DOI
- Zhang, J., Wu, J., Li, H., Chen, Q., & Lin, J. M. (2015). An in vitro liver model on microfluidic device for analysis of capecitabine metabolite using mass spectrometer as detector. Biosensors & Bioelectronics, 68, 322–328. DOI
- Li, Z., Su, W., Zhu, Y., Tao, T., Li, D., Peng, X., & Qin, J. (2017). Drug absorption related nephrotoxicity assessment on an intestine-kidney chip. Biomicrofluidics, 11(3), 034114. DOI
- Sung J. H. (2020). A body-on-a-chip (BOC) system for studying gut-liver interaction. Methods in Cell Biology, 158, 1–10. DOI
- Picollet-D'hahan, N., Zuchowska, A., Lemeunier, I., & Le Gac, S. (2021). Multiorgan-on-a-Chip: A Systemic Approach To Model and Decipher Inter-Organ Communication. Trends in Biotechnology. Advance online publication. DOI
- An, F., Qu, Y., Luo, Y., Fang, N., Liu, Y., Gao, Z., Zhao, W., & Lin, B. (2016). A Laminated Microfluidic Device for Comprehensive Preclinical Testing in the Drug ADME Process. Scientific Reports, 6, 25022. DOI
- Esch, M. B., Mahler, G. J., Stokol, T., & Shuler, M. L. (2014). Body-on-a-chip simulation with gastrointestinal tract and liver tissues suggests that ingested nanoparticles have the potential to cause liver injury. Lab on a Chip, 14(16), 3081–3092. DOI
- Sung, J. H., Esch, M. B., Prot, J. M., Long, C. J., Smith, A., Hickman, J. J., & Shuler, M. L. (2013). Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab on a Chip, 13(7), 1201–1212. DOI
- Ekkers, D. M., Branco Dos Santos, F., Mallon, C. A., Bruggeman, F., & van Doorn, G. S. (2020). The omnistat: A flexible continuous-culture system for prolonged experimental evolution. Methods in Ecology and Evolution, 11(8), 932–942. DOI
- Liu, Y., El Masoudi, A., Pronk, J. T., & van Gulik, W. M. (2019). Quantitative Physiology of Non-Energy-Limited Retentostat Cultures of Saccharomyces cerevisiae at Near-Zero Specific Growth Rates. Applied and Environmental Microbiology, 85(20), e01161-19. DOI
- Holt, L. J., Hallatschek, O., & Delarue, M. (2018). Mechano-chemostats to study the effects of compressive stress on yeast. Methods in Cell Biology, 147, 215–231. DOI
- Ekkers, D. M., Branco Dos Santos, F., Mallon, C. A., Bruggeman, F., & van Doorn, G. S. (2020). The omnistat: A flexible continuous-culture system for prolonged experimental evolution. Methods in Ecology and Evolution, 11(8), 932–942. DOI
- Spence, J. R., Mayhew, C. N., Rankin, S. A., Kuhar, M. F., Vallance, J. E., Tolle, K., Hoskins, E. E., Kalinichenko, V. V., Wells, S. I., Zorn, A. M., Shroyer, N. F., & Wells, J. M. (2011). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature, 470(7332), 105–109. DOI
- Hill, D. R., Huang, S., Nagy, M. S., Yadagiri, V. K., Fields, C., Mukherjee, D., Bons, B., Dedhia, P. H., Chin, A. M., Tsai, Y. H., Thodla, S., Schmidt, T. M., Walk, S., Young, V. B., & Spence, J. R. (2017). Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. eLife, 6, e29132. DOI
- Kolawole, A. O., & Wobus, C. E. (2020). Gastrointestinal organoid technology advances studies of enteric virus biology. PLoS pathogens, 16(1), e1008212. DOI
- Barrila, J., Crabbé, A., Yang, J., Franco, K., Nydam, S. D., Forsyth, R. J., Davis, R. R., Gangaraju, S., Ott, C. M., Coyne, C. B., Bissell, M. J., & Nickerson, C. A. (2018). Modeling host-pathogen interactions in the context of the microenvironment: three-dimensional cell culture comes of age. Infection and Immunity, 86(11), e00282-18. DOI
- Pusch, J., Votteler, M., Göhler, S., Engl, J., Hampel, M., Walles, H., & Schenke-Layland, K. (2011). The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials, 32(30), 7469–7478. DOI
- Ingber D. E. (2016). Reverse Engineering Human Pathophysiology with Organs-on-Chips. Cell, 164(6), 1105–1109. DOI
- Imura, Y., Asano, Y., Sato, K., & Yoshimura, E. (2009). A microfluidic system to evaluate intestinal absorption. Analytical Sciences: the International Journal of the Japan Society for Analytical Chemistry, 25(12), 1403–1407. DOI
- Shah, P., Fritz, J. V., Glaab, E., Desai, M. S., Greenhalgh, K., Frachet, A., Niegowska, M., Estes, M., Jäger, C., Seguin-Devaux, C., Zenhausern, F., & Wilmes, P. (2016). A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nature communications, 7, 11535. DOI
- Eain, M. M. G., Baginska, J., Greenhalgh, K., Fritz, J. V., Zenhausern, F., & Wilmes, P. (2017). Engineering solutions for representative models of the gastrointestinal human-microbe interface. Engineering, 3(1), 60-65. DOI
- Jalili-Firoozinezhad, S., Gazzaniga, F. S., Calamari, E. L., Camacho, D. M., Fadel, C. W., Bein, A., Swenor, B., Nestor, B., Cronce, M. J., Tovaglieri, A., Levy, O., Gregory, K. E., Breault, D. T., Cabral, J., Kasper, D. L., Novak, R., & Ingber, D. E. (2019). A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nature Biomedical Engineering, 3(7), 520–531. DOI