|
Особенности экспрессии и выделения укороченной рекомбинантной реналазы в прокариотических клетках Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121 Москва, Погодинская ул., д.10; * e-mail: valfed38@yandex.ru Ключевые слова: реналаза; экспрессия белка; тельца включения; растворение белков; выделение DOI: 10.18097/BMCRM00158 ВВЕДЕНИЕ
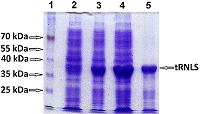
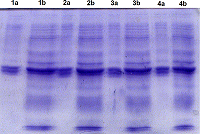
Реналаза (RNLS)– флавопротеин, N-концевой пептид которого (аминокислотные остатки (а.о.) 1-17 полипептидной цепи) выполняет несколько важных функций [1-3]. Он участвует в формировании так называемой укладки Россмана (2-35 а.о.), необходимой для «размещения» кофактора FAD в третичной структуре этого белка [3]. Такая полноразмерная RNLS функционирует как FAD-зависимая оксидоредуктаза (КФ 1.6.3.5) [4]. При секреции RNLS во внеклеточное пространство этот пептид отщепляется и образующаяся укороченная внеклеточная RNLS не может связывать FAD [3, 5] и выполняет различные некаталитические функции [6-9]. Для их изучения используют различные подходы, предусматривающие химический синтез пептидов, воспроизводящих определенные аминокислотные последовательности RNLS [10], а также создание химерного гена, кодирующего RNLS с модифицированным N-концом [11]. Используя экзоновый метод сборки кодирующих нуклеотидных последовательностей [12], были получены генетические конструкции, содержащие ген полноразмерной RNLS человека и кодирующую нуклеотидную последовательность RNLS человека, лишенную N-концевого сигнального пептида (tRNLS) [11], а также осуществлена экспрессия полноразмерной RNLS в бактериальных [12] и эукариотических клетках [5]. В данной работе мы осуществили экспрессию генетической конструкции, кодирующей tRNLS с С-концевой гексагистидиновой последовательностью, в бактериальных клетках E. coli Rosetta (DE3). Как и в случае полноразмерной RNLS, рекомбинантная tRNLS накапливается в тельцах включения в нерастворимой форме, которая может быть переведена в растворимую форму в присутствии высокой концентрации мочевины или гуанидинхлорида. Однако в отличие от полноразмерной RNLS, которая солюбилизировалась в присутствии 8 М мочевины, более эффективная солюбилизация tRNLS была достигнута в присутствии 6 М гуанидинхлорида. МАТЕРИАЛЫ И МЕТОДЫ В работе были использованы следующие реагенты: канамицин сульфат, додецилсульфат натрия (SDS) («Serva», Германия); изопропил-b-D-тиогалактопиранозид (ИПТГ) («ThermoScientific», США); гуанидинхлорид (GdnHCl) и мочевина («Amresco», США); Ni-сефароза («GE Healthcare», Швеция); мочевина («Amresco», США); Остальные химические реактивы были приобретены у («Sigma», США). Клетки E. coli штамм Rosetta (DE3) были приобретены у «Novagen» (Великобритания). Электрофорез Электрофорез в 12% полиакриламидном геле (ПААГ) в присутствии SDS(SDS-PAGE) проводили в системе Лэммли [13]. Трансформация клеток E. coli штамм Rosetta (DE3) плазмидными векторами Плазмидными векторами pET-hRen и pET-hRen(-sp), полученные нами ранее [12], трансформировали клетки E. coli штамм Rosetta (DE3) согласно протоколу [14, 15], используя 1 мкл вектора. Трансформированные клетки высевали на чашки с LB-агаром, содержащим канамицин (50 мкг/мл), необходимый для селекции трансформированных клеток. После ночи инкубации при 37°C отдельные колонии инокулировали в 4 мл LB-среды, содержащей 50 мкг/мл Км и инкубировали в течение ночи при 37°C. Наработка биомассы клетокE. coli, содержащих рекомбинантный белок RNLS Затравочную культуруклеток E. coli штамм Rosetta (DE3) использовали для инокуляции 50 мл LB-среды, содержащей канамицин (50 мкг/мл). Затравочную культуру выращивали 12 ч в термостатированном шейкере Ecotron («Infors», Швейцария) при частоте вращения 180 об/мин и температуре 37°C. Для наработки биомассы культуру клеток E. coli штамм Rosetta (DE3) высевали в 5 л колбу, содержащую 1 л LB-среды с канамицином (50 мкг/мл), и продолжали культивирование в прежних условиях до достижения значений оптической плотности при длине волны 600 нм (ОП600) 0.6 ед.(1.5-2 ч). Затем вносили ИПТГдо конечной концентрации 1 мМ и продолжали культивирование в прежних условиях, примерно 3.5 ч. Полученную биомассу (примерно 1.5 г) осаждали в центрифуге AvantiJ-Ec ротором JA-14 («BeckmanCoulter», США) 10 мин при скорости 4000 об/мин и температуре 4°C. При необходимости биомассу хранили при -20°C. Присутствие целевого продукта оценивали в лизате целых клеток с помощью 12% SDS-PAGE и визуализации с помощью окрашивания Кумасси R-250. Получение телец включения (ТВ) Осадок бактериальной биомассы (1.5 г), содержащий рекомбинантный белок, ресуспендировали в литическом буфере (фосфатно-солевой буфер: 5 мМ ЭДТА, 7 мкМ β-меркаптоэтанол). Суспензию подвергали ультразвуковой дезинтеграции на приборе 130-Watt Ultrasonic Processor («Cole-Parmer», США) при 130 Вт и 20 кГц трижды по 30 с с интервалом 90 с во льду. Затем проводили осаждение в центрифуге AvantiJ-Ec ротором JA-20 («Beckman Coulter») 20 мин при 10000 об/мин и температуре 4°C. Супернатант удаляли, полученный осадок растворяли в буфере для ферментативной обработки (10 мМ NaCl, 6 мМ MgCl2, 10 мМ CaCl2, 40 мМ Tris-HCl, pH 8.0), а затем добавляли 6 мкл РНКазы-А (10 мг/л) и 15 мкл ДНКазы-I (10 мг/мл). Суспензию инкубировали 15 мин при температуре 37°C. Повторную обработку суспензии ультразвуком и ее последующее центрифугирование проводили как описано выше. Супернатант удаляли, а полученный осадок ресуспендировали в промывочном буфере (фосфатно-солевой буфер: 5 мМ ЭДТА, 20% сахароза, 1% Triton X-100) и инкубировали 10 мин при температуре 4°C, а затем центрифугировали, как описано выше. Супернатант удаляли и собирали осадок, содержащий ТВ. Для 1.5 г.биомассы, полученной из 1 л культуральной среды, использовали по 30 мл всех растворов. При необходимости осадок ТВ хранили при -20°C. Выделение рекомбинантнойt RNLS Для выделения рекомбинантного белка ТВ солюбилизировали при помощи буфера А, содержащего 8 М мочевину, 0.1 М NaH2PO4, 0.01 М Tris-HCl, рН 8.0. или буфера В, содержащего 6 М GdnHCl, 0.1 М NaH2PO4, 0.01 М Tris-HCl, рН 8.0. Препараты ТВ, полученные из 1 л культуральной среды, обрабатывали 100 мл соответствующего буфера. Солюбилизацию рекомбинантного белка из ТВ осуществляли в течение суток при комнатной температуре при 120-160 об/мин, используя шейкер Ecotron. Нерастворившиеся компоненты ТВ удаляли центрифугированием (центрифуга AvantiJ-Ec ротором JA-20 («Beckman Coulter») при 10000 об/мин и комнатной температуре дважды по 30 мин. К осветленному раствору, полученному после центрифугирования, добавляли 2 мл суспензии Ni-Sepharose. Смесь инкубировали 1 ч при комнатной температуре в шейкере Ecotron при частоте вращения 40 об/мин. После этого раствор переносили в колонку для хроматографии и проводили хроматографию с использованием системы BiologicLP («Bio-Rad», Германия). Перед хроматографированием колонку промывали 200 мл буфера С, содержащего 8 М мочевину, 0.1 М NaH2PO4, 0.01 М Tris-HCl, рН 6.3, а затем 50 мл буфером D, содержащим 8 М мочевину, 0.1 М NaH2PO4, 0.01 М Tris-HCl, рН5.9. Рекомбинантный белок с колонки элюировали буфером E, содержащим 8 М мочевину 0.1 М NaH2PO4, 0.01 М Tris-HCl, рН 4.5. Белок собирали при пиковом значении хромотограммы. Рефолдинг собранного рекомбинантного белка осуществляли с помощью ступенчатого диализа против нескольких перемен этого же буферного раствора с понижение концентрации мочевины от 8 М до 6 М, 4 М, 2 М и 1 М, а затем в буфере без мочевины (50 мМ Tris-HCl, рН 9.0). Диализ проводили при температуре 4°С. Концентрацию белка определяли спектрофотометрически (Genesys 6, «ThermoScientific», США) при длине волны 280 нм. При расчете концентрации рекомбинантных белков использовали коэффициент экстинкции 0.98, который был рассчитан в программе OMIGA. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Для изучения свойств полноразмерной RNLS и RNLS, лишенной N-концевого сигнального пептида (tRNLS), нами были сконструированы вектора для их экспрессии в бактериальной системы [11, 12, 16]. Обычно при экспрессии рекомбинантных белков клетках E. coli белки нарабатываются в избыточном количестве и образуют нерастворимые агрегаты, известные как ТВ. [17, 18]. Ранее в клетках E. coli штамм Rosetta (DE3) нами был экспрессирован полноразмерный белок RNLS, целевой продукт которой также накапливался именно в виде ТВ [12, 19]. При разрушении клеток ТВ легко можно отделить от других компонентов клеток. Однако дезагрегация ТВ без необратимой денатурации белка является трудоемкой задачей. Поскольку ТВ содержат относительно чистые и интактные рекомбинантные белки, существует несколько подходов очистки рекомбинантных белков. Обычно рекомбинантные белки образуют агрегированные формы, поэтому они денатурируются и растворяются при высокой концентрацией денатурирующих агентов, таких как мочевина, GdnHCl или ионные детергенты, такие как N-лауроилсаркозин или SDS. Эти химические реагенты применяют для уменьшения нековалентных взаимодействий между молекулами белка. Кроме того, добавляют дитиотреитол или 2-меркаптоэтанол для уменьшения образования нежелательных наружных или внутри молекулярных дисульфидных связей. Рефолдинг денатурированных белков (перевод развернутой формы белка в активные белки) происходит за счет удаления денатуранта. Эффективность рефолдинга оценивают по биологической активности данного белка, например, по его ферментативной активности. Процедура удаления денатуранта из денатурированных белков является ключевым этапом для эффективного восстановления активности белка. Используют различные подходы к переводу неактивных белков в активный, такие как хроматографические или нехроматографические стратегии [20-22]. Для рефолдинга tRNLS мы воспользовались методам ступенчатого диализа. При диализе химически денатурированный белок повторно укладывается в свою нативную третичную структуру. Уменьшение концентрации денатурирующего агента приводит к постепенной укладке третичной структуры белка. Ступенчатый диализ способствует медленному уменьшению концентрации денатурирующего агента и, в конечном итоге, приводит к хорошему выходу рефолдинга, т.е. не приводит к неправильной укладке и агрегации белков из-за уменьшения контакта между открытыми гидрофобными поверхностями [23, 24]. Ранее препаративные количества полноразмерной рекомбинантной RNLS были получены из ТВ, которые солюбилизировали в течение ночи при помощи буферного раствора, содержащего 8 М мочевину, с последующей хроматографией на Ni- сефарозе [12]. При экспрессии белка tRNLS (лишенной N-концевой сигнальной последовательности) в клетках Е. coli штамма Rosetta (DE3) (вектор pET-hRen(-sp)) также успешно происходило формирование ТВ, в которых накапливался целевой белок (рис. 1, дорожки 3 и 4).
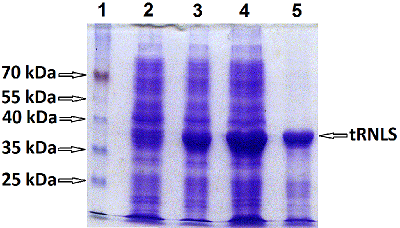
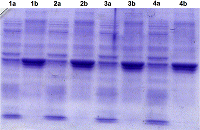
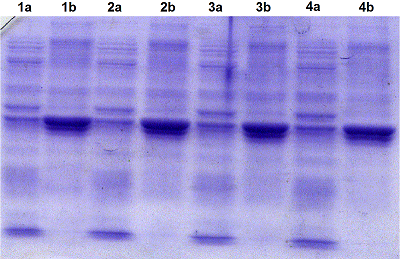
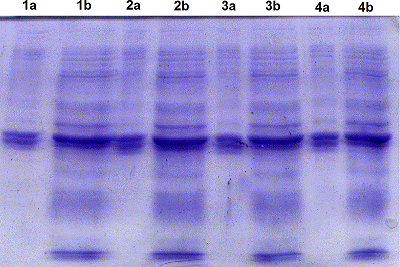
Однако при использовании буфера, содержащего 8 М мочевину (буфер А) рекомбинантный белок укороченной RNLS (tRNLS) не переводился из ТВ в растворимую форм (рис. 2). Перевод tRNLS из ТВ в растворимую форму достигался при использовании 6М GdnHCl (рис. 3). Для определения растворимости tRNLS в ТВ исследовали растворение белка в 8 М буферном растворе мочевины (буфер А) и в 6 М буферном растворе GdnHCl (буфер В). Аликвоты ТВ, содержащие буферы А и В, инкубировали 15 мин, 30 мин, 1 ч и 12 ч в термостатированном шейкере Ecotron при 180 об/мин и температуре 37ºC. После инкубации каждый препарат центрифугировали и отделяли супернатант и осадок. Полученные осадки растворяли в буфере А, объем которого был равен супернатантам и анализировали при помощи SDS-PAGE. Оказалось, что инкубация ТВ в присутствии 8 М мочевины не приводила к растворению целевого рекомбинантного продукта даже после 12-часовой инкубации. Практически весь рекомбинантый белок tRNLS оставался в осадке (Рис.2). В то же время использование 6 М GdnHCl способствовало растворению значительной части рекомбинантного белка уже в течение 15 мин (рис. 3, дорожки 1a и 1b), а в течение 12 ч происходило растворение большей его части (рис.3, дорожки 4a и 4b). Для полного перевода RNLS в растворимую форму инкубацию в буфере В осуществляли в течение суток в шейкере Ecotron при частоте вращения 120-160 об/мин при комнатной температуре. Растворимую форму tRNLS осветляли дважды центрифугированием и добавляли суспензию Ni-Sepharose. Полученный раствор переносили в колонку для хроматографии и проводили хроматографию. С колонки рекомбинантный белок элюировали при рН 4.5 буфером E, содержащим 8 М мочевину. Рефолдинг химерного белка tRNLS, полученного после хроматографии, осуществляли при помощи ступенчатого диализа с постепенным понижением концентрации мочевины до 6 М, затем до 4 М, потом до 2 М и до 1 М мочевины при рН 9.0. На последнем этапе диализ проводили в растворе (50 мМTris-HCl, рН 9.0), не содержащем мочевину (см. МАТЕРИАЛЫ И МЕТОДЫ). Следует отметить, что при концентрировании рекомбинантного белка из разбавленных растворов, в которых проводится рефолдинг, белок часто вновь выпадает в осадок.
ЗАКЛЮЧЕНИЕ Экспрессия рекомбинантных RNLS в клетках E. Coli Rosetta (DE3) приводит к накоплению целевого продукта в ТВ в нерастворимой форме, перевод которого в растворимую форму для последующей хроматографической очистки на Ni-сефарозе может быть достигнут при использовании высоких (молярных) концентраций мочевины или GdnHCl. При этом, в отличие от полноразмерной RNLS, которая солюбилизировалась в присутствии 8 М мочевины (буфер А), более эффективная солюбилизация tRNLS была достигнута в присутствии 6 М GdnHCl (буфер В). После хроматографии на Ni-Sepharose в присутствии 8 М мочевины рефолдинг как RNLS, так и tRNLS может быть достигнут при помощи диализа при рН 9.0. с постепенным понижением концентрации мочевины. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Данная работа не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов. ФИНАНСИРОВАНИЕ Исследования выполнены в рамках проекта РФФИ № 20–015–00104. КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|