|
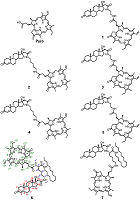
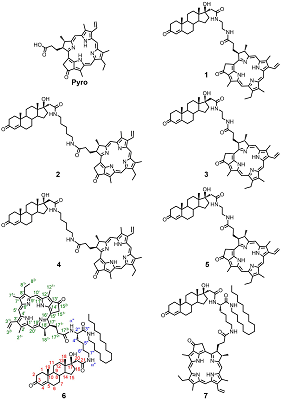
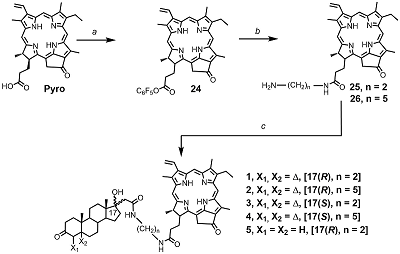
Conjugates of Pyropheophorbide a with 17-Substituted Steroidal Androgens. Synthesis, Molecular Modeling, Interaction with Some Cancer Cells* 1V.N. Orekhovich Institute of Biomedical Chemistry, 10 Pogodinskaya str., 119121, Moscow, Russia; 2RUDN University, 6 Miklukho-Maklaya street, 117198, Moscow, Russia Key words: steroid conjugates; chemical synthesis; tetrapyrrolic macrocycles; molecular models; prostate carcinoma cells; phospholipid micelles. DOI: 10.18097/BMCRM00167 INTRODUCTION Tetrapyrrolic macrocycles porphyrins and chlorins have wide range of biomedical applications owing to their unique photochemical and photophysical properties. They are used in optical imaging, fluorescent labeling, photodynamic inactivation of microbial infections, and photodynamic therapy of solid tumors. Coupling of macrocycles with drugs or fragments of biological active molecules significantly improves delivery and distribution of macrocycle-based compounds to a specific location within the cells, facilitates their transport via receptor or drug mediated endocytosis, increased their specificity and selectivity, affects their photo chemical properties and biological activity [1-7]. Conjugation of pheophorbide a or pyropheophorbide a with steroids is considered to be a promising approach for development of new bifunctional constructs possessing enhanced delivery to specific targets [8-11]. Specifically, such conjugates with estradiol were efficiently internalized by estrogen receptor positive cells, accumulated in nuclei, and revealed a potency of application as sensitizer for photodynamic therapy of breast cancer [8-11]. In this study we have synthesized and investigated seven new conjugates of pyropheophorbide a (Pyro) with androgen receptor ligands – testosterone, dihydrotestosterone and epitestosterone 1–7 (Figure 1). In bifunctional conjugates 1–5 macrocyclic and steroidal moieties are connected with ethylene diamine or 1,5-diamino pentane linkers; in conjugates 6 and 7 (L)-lysine residue was used as a linker. The latter allowed to introduce an additional functional fragment, the lipophylic hexadecyl chain, permitting simple solubilization of conjugates in an aqueous medium in the form of mixed micelles with phosphatidyl choline [12].
Presented here results of studies of spectral properties and molecular models of conjugates 1–7 revealed a significant influence of the structure on the conformation of conjugates. The study of interaction of conjugates with prostate carcinoma LNCaP and PC-3 cells indicated that conjugates 1–5 were efficiently uptaken and internalized by cells and potently inhibited their growth and proliferation. Anti-proliferative activity of conjugates 1–5, as well as their photo induced toxicity in prostate carcinoma cells depended on the length of linker and the stereochemical configuration of C17 atom in steroidal part. On the other hand, conjugates 6 and 7 were insoluble in aqueous medium, they did not bind to prostate carcinoma LNCaP and PC-3 cells and did not affect the growth and proliferation of these cells. Nevertheless, they may be uptaken by human hepatocarcinoma Hep G2 cells in the form of mixed micelles with phosphatidyl choline or with biocompatible detergent. The presented data showed that some of new synthesized conjugates of pyropheophorbide a with steroids could be considered as prospective agents for various biomedical experiments in cultured cells. MATERIALS AND METHODS
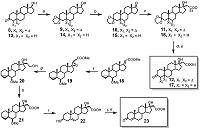
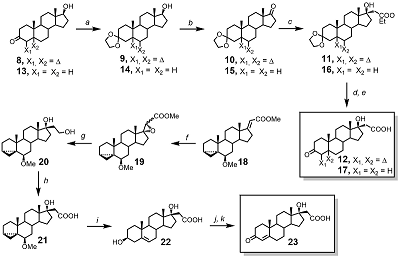
Materials and general methods HRMS were registered using Bruker ‘Apex Ultra’ FT ICR MS and Bruker ‘Daltonics micrOTOF-Q II’ instruments at the ion positive electrospray ionization mode; 1H NMR and 13C NMR spectra – on an AMX-III instrument (“Bruker”, 400 MHz) in CDCl3 (signals of 1H in CHCl3 was 7.28 ppm, and 13C in CDCl3 was 77.16 ppm). Assignment of ambiguous proton resonances in target compounds was performed by analyzing the set of 2D NMR spectra (data not shown). Absorption spectra were measured with a “Cary Spectra 100” spectro-photometer in CHCl3 and CH2Cl2 using a quartz cell with the 1 mm optical path length; particle size distribution was measured using “DelsaNano Beckman Coulter” instrument. Flash chromatography was performed on silica gel (0.035–0.070 mm) from “Acros”, TLC – on HPTLC Silica gel F254 GLP 105564 glass plates from ‘Merck’; compounds on the plates were visualized by UV light (Filter 254 nm); and/or by spraying the dried developed plates with 5% (NH4)2MoO4 in 10% sulfuric acid, followed by heating; pyropheophorbide a derivatives were visible on the plates without any treatments. Dihydrotestosterone, testosterone, N-hydroxysuccinimide, dicyclohexyl carbodiimide, 4-dimethylaminopyridine, N(α)-Fmoc-N(ε)-Boc-Lys, ethylene diamine, 1,5-diamino pentane were obtained from “Acros”; soya bean PC “Lipoid S-100” was purchased from “Lipoids” (Germany), pluronic F68 – from “BASF” (Germany). Pyro was prepared from methyl pheophorbide a according to procedure [25]; methyl [17(20)E]-6β-methoxy-3α,5α-cyclopregn-17(20)-en-21-oate 18 was synthesized according to procedure [17]; Dess-Martin periodinane – according to procedure [14], other reagents and solvents were purchased from “Aldrich” (USA), “Merck” (Germany), “Acros”, “Fluka” (Switzerland) and “Spectra Chem (Moscow, Russia)”. Synthesis of compounds 9 – 12 and 14 – 17 is presented in the “Supplementary data” section. Chemical synthesis Methyl 6β-methoxy-17α,20(R,S)-epoxy-3α,5α-cyclopregn-17(20)-en-21-oate (19) m-Chloroperbenzoic acid (2.06 g, 8.37 mmol) was added to the solution of compound 18 (2.00 g, 5.58 mmol) in CH2Cl2 (60 mL) and the mixture was stirred and heated under re-flux for 8 h, disappearance of starting compound was monitored by TLC. The saturated solutions of NaHCO3 (70 mL) and NaHSO3 (70 mL) were added; resulted mixture was vigorously stirred for 30 min, then the layers were separated. The aqueous layer was then extracted with dichloromethane (2×30 mL). The combined extract was washed with brine (40 mL), dried over Na2SO4, and evaporated. The residue was purified by silica gel flash chromatography in hexane – EtOAc (8:1) followed by evaporation to obtain epoxide 19 (the mixture of two isomers in a ratio of 3:1, 1.34 g, 3.57 mmol, 64%) as colorless glass. HRMS, calculated for [C23H35O4]+: 375.2530; found: 375.2529. 1H NMR for major isomer: 0.44 and 0.65 (each 1H, m, H-4); 0.93 (3H, s, H-18); 1.00 (3H, s, H-19); 2.78 (1H, t, J=2.7 Hz, H-6); 3.32 (3H, s, CH3OC6); 3.45 (1H, s, H-20); 3.75 (3H, s, CH3OC21); 1H NMR for minor isomer: 0.44 and 0.65 (each 1H, m, H-4); 0.89 (3H, s, H-18); 1.02 (3H, s, H-19); 2.78 (1H, t, J=2.7 Hz, H-6); 3.32 (3H, s, CH3OC6); 3.36 (1H, s, H-20); 3.75 (3H, s, CH3OC21). 6β-Methoxy-17α,21-dihydroxy-3α,5α-cyclopregnane (20) The solution of compound 19 (780 mg, 2.1 mmol) in abs. THF (20 mL) was added by drops to the stirred suspension of LiAlH4 (175 mg, 4.6 mmol) in abs. THF (40 mL), and the mixture was stirred and heated under reflux for 2 h. After cooling, excess of LiAlH4 was decomposed by adding of ice water. The mixture was filtered, the residue was washed with Et2O (2×30 mL). The combined extract was dried over Na2SO4, and evaporated to obtain diol 20 (680 mg, 1.9 mmol, 90%) as colorless glass. HRMS, calculated for [C22H37O3]+: 349.2737; found: 349.2732; 1H NMR: 0.43 and 0.64 (each 1H, m, H-4); 0.74 (3H, s, H-18); 1.03 (3H, s, H-19); 2.76 (1H, m, H-6); 3.32 (3H, s, CH3O); 3.90 (2H, m, H-21); 13C NMR: 13.2, 16.0, 19.4, 21.6, 22.4, 23.7, 25.0, 29.8, 30.8, 30.9, 33.5, 35.3, 36.8(×2), 43.6, 47.5, 48.1, 49.9, 56.7, 61.1, 82.4, 85.5. 6β-Methoxy-17α-hydroxy-3α,5α-cyclopregnan-21-oic acid (21) Diol 20 (2.23 g, 6.4 mmol) was dissolved in acetone (120 mL), then KBrO3 (6.4 g, 38.5 mmol), water (80 mL), and RuO2xH2O (10 mg) were added, and the mixture was heated under reflux for 20 min, followed by cooling to room temperature. Thereafter EtOH (25 mL) was added by drops, the mixture was filtered, the residue was washed with acetone. The combined filtrate was evaporated, the residue was treated with CHCl3 (150 mL) and water (50 mL). The chloroform extract was washed with brine (50 mL), dried over Na2SO4, and evaporated. The residue was applied on the top a silica gel column; the column was initially washed with hexane – EtOAc (2:1) to remove byproducts, then the target compound was eluted with hexane – EtOAc – CH3COOH (50:49:1) to obtain acid 21 (1.55 g, 4.3 mmol, 67%) as white foam. HRMS, calculated for [C22H35O4]+: 363.2530; found: 363.2532; 1H NMR: 0.43 and 0.64 (each 1H, m, H-4); 0.76 (3H, s, H-18); 1.01 (3H, s, H-19); 2.58 (2H, AB system, H-20); 2.78 (1H, m, H-6); 3.32 (3H, s, CH3O); 13C NMR: 13.2, 16.0, 19.7, 21.6, 22.2, 23.7, 25.0, 29.8, 30.8, 33.5, 35.2, 35.3, 37.5, 39.8, 43.5, 47.5, 47.8, 49.6, 56.6, 81.8, 82.5, 178.1. 3β,17α-Dihydroxypregn-5-en-21-oic acid (22) Compound 21 (350 mg, 0.97 mmol) was dissolved in THF (15 mL) and after addition of 15% aqueous H2SO4 (4 mL) the mixture was heated under reflux for 20 min. After cooling water (50 mL) was added and the mixture was extracted with CHCl3 (3×50 mL). The combined chloroform extract was washed with brine (30 mL), dried over Na2SO4, and evaporated to obtain acid 22 (300, mg, 0.86 mmol, 89%) as white powder which was used without purification. The analytical sample was obtained after silica gel flash chromatography in hexane – EtOAc – CH3COOH (75:24:1). HRMS, calculated for [C21H33O4]+: 349.2373; found: 349.2370; 1H NMR: 0.56 (3H, s, H-18); 0.84 (3H, s, H-19); 2.42 (2H, AB system, H-20); 3.38 (1H, m, H-3); 5.176 (1H, m, H-6); 13C NMR: 14.91, 18.65, 20.37, 23.51, 29.90; 30.49, 31.68, 32.05, 36.28, 36.57, 36.97, 39.42; 41.12, 46.90, 49.45, 49.74; 71.55; 82.16; 121.36; 140.35; 178.21. 3-Oxo-17α-hydroxypregn-4-en-21-oic acid (23) Dess-Martin periodinane (902 mg, 2.12 mmol) was added to the stirred suspension of compound 22 (300 mg, 0.86 mmol) in dichloromethane (20 mL) and after addition of water (10 μL, 0.56 mmol) the mixture was stirred for 30 min more; disappearance of compound 22 during the reaction was controlled by TLC. The mixture was cooled to 4°C, and after dropwise addition of EtOH (20 mL) it was poured into water (50 mL) and extracted with CHCl3 (3×20 mL). The extract was washed with brine (30 mL), dried over Na2SO4, and evaporated. The residue dissolved in abs. EtOH (8 mL) was mixed with oxalic acid (36 mg, 0.4 mmol). The mixture was stirred and heating under reflux for 10 min and poured into water (50 mL). The suspension obtained was extracted with CHCl3 (3×20 mL). The extract was washed with brine (30 mL), dried over Na2SO4 and evaporated. The residue was purified by silica gel flash chromatography in hexane – acetone – CH3COOH (64:35:1) and evaporated to obtain 3-oxo-17α-hydroxypregn-4-en-21-oic acid 23 (193 mg, 0.56 mmol, 65%) as white solid. HRMS, calculated for [C21H31O4]+: 347.2217; found: 347.2219; 1H NMR: 0.76 (3H, s, H-18); 1.18 (3H, s, H-19); 2.59 (2H, AB system, H-20); 3.38 (1H, br. s, 17-OH); 5.73 (1H, s, H-4); 13C NMR: 15.5, 17.4, 20.6, 23.7; 30.2, 32.0, 32.9, 33.9, 35.7, 35.9, 37.4, 38.7, 39.6, 47.1, 49.0, 53.5, 81.5, 123.8, 171.8, 177.1, 200.0. Pentafluorophenyl pyropheophorbide a (24) Pyropheophorbide a (Pyro) (150 mg, 0.28 mmol) was dissolved in 15 mL of CH2Cl2, then pentafluorophenyl trifluoroacetate (0.096 mL, 0.56 mmol) was added, thereafter Et3N (0.039 mL, 2.8 mmol) was added dropvise to stirred solution during 10 min. The formation of pentafluorophenyl ester was controlled by TLC. After the reaction was completed, the solvent was evaporated in vacuo, the residue was twice evaporated with toluene, and purified by chromatography on silica gel in hexane – acetone (4:1) to give ester 24 (186 mg, 2.7 mmol, 95 %). HRMS, calculated for [C39H34F5N4O3]+: 701.2551; found: 701.2554. 1H NMR: -1.45 (1H br.s, N-H); 1.68 (1H, t, J = 7.6 Hz, 82-H); 1.84 (3H, d, J = 7.3 Hz, 18-CH3); 3.22, 3.41, 3.65 (each 3H, s, 2-, 7-, 12-CH3); 4.37, 4.52 (each 1H, m, 171-H and 81-H); 5.18, 5.24 (each 1H, d, J = 19.7 Hz, 172-H), 6.17 (1H, dd, J = 11.5 Hz and J = 1.4 Hz, 82-H, cis), 6.22 (1H, dd, J = 17.9 Hz and J = 1.4 Hz, 82-H, trans), 7.98 (1H, dd, J = 11.5 Hz and J = 17.9 Hz, 81-H), 8.57, 9.37, 9.48 (each 1H, s, 5-, 10-, 20-H). 13C NMR: 11.28; 12.12; 17.44; 19.53; 23.14; 29.72; 30.32; 47.99; 50.01; 51.41; 93.05; 97.41; 104.40; 105.98; 122.76; 128.57; 129.23; 130.37; 131.83; 136.17; 136.27; 136.54; 136.71; 136.83; 137.99; 138.38; 139.25; 139.92; 140.90; 141.92; 142.47; 145.27; 149.23; 151.02; 155.65; 159.57; 169.17; 177.30; 196.55; Absorption spectra in CH2Cl2, λmax, nm: 398, 498, 660. Procedure for preparation of amides 25 and 26 The mixture of C6F5-Pyro 25 (210 mg, 0.3 mmol), diamine (ethylene diamine or 1,5-diaminopentane, 6.0 mmol) and abs. CH2Cl2 (10 mL) was stirred for 2 h, then the mixture was poured into 0.1M CH3COONa buffer, pH 5 (20 mL), extracted with CH2Cl2 (2×20 mL), the combined extract was washed with brine (20 mL), dried over Na2SO4, and evaporated. Then the residue was dissolved in tetrahydrofuran (30 mL), the solution was dried over granulated KOH, followed by evaporation to dryness. 173[(2-Aminoethyl)-amido]-pyropheophorbide a (25) Compound 25 was purified by flash chromatography in CHCl3 – MeOH – NH4OH (90:9:1) mixture, was obtained as black amorphous powder (135 mg, 0.22 μmol, 73%). HRMS, calculated for [C35H41N6O2]+: 577.3291, found: 577.3292; 1H-NMR: -1.70, 0.33 (each 1H, br.s, N-H); 1.62 (3H, t, J = 7.6 Hz, H-82’); 1.75 (3H, d, J = 7.3 Hz, 18-CH3); 3.18, 3.37, 3.41 (each 3H, s, H-2’, H-7’, H-12’); 4.23, 4.45 (each 1H, m, H-171’ and H-81’); 4.98, 5.19 (each 1H, d, J = 19.7 Hz, H-172’); 6.13 (1H, dd, J = 11.5 Hz and J = 1.4 Hz, H-32’, trans); 6.24 (1H, dd, J = 17.9 Hz and J = 1.4 Hz, H-32’, cis); 7.95 (1H, dd, J = 11.5 Hz and J = 17.9 Hz, H-31’); 8.50, 9.24, 9.30 (each 1H, s, H-5’, H-10’, H-20’); 13C-NMR: 11.2; 11.8; 12.1; 17.4; 19.4; 23.0; 28.3; 30.2; 30.9; 32.8; 40.9; 41.7; 48.0; 47.0; 51.7; 92.9; 97.1; 103.9; 106.0; 122.7; 128.1; 129.2; 131.5; 135.8; 136.0; 136.1; 137.7; 144.9; 148.9; 150.7; 155.1; 160.4; 171.7; 172.4; 196.1. Absorption spectra in CH2Cl2, λmax, nm (ε): 413 (85 000); 507 (8 900); 538 (8 000); 609 (7 000); 665 (35 200). 173[(2-Aminoethyl)-amido]-pyropheophorbide a (26) Compound 26 was purified by flash chromatography in CHCl3 – MeOH – NH4OH (90:9:1) mixture, was obtained as black amorphous powder (130 mg, 0.21 μmol, 70%). HRMS, calculated for [C38H47N6O2]+: 619.3760; found: 619.3749; 1H NMR: −1.70, 0.40 (each 1H, br.s, N–H); 1.64 (3H, t, J=7.6 Hz, H-82’); 1.78 (3H, d, J=7.3 Hz, H-181’); 3.20, 3.38, 3.46 (each 3H, s, H-21’, H-71’, H-121’); 4.30, 4.48 (each 1H, m, H-171’ and H-81’); 5.05 (1H, br. s, NH-CO); 5.05, 5.21 (each 1H, d, J=19.7 Hz, H-151’); 6.15 (1H, dd, J=11.5 Hz and J=1.4 Hz, H-32’, trans); 6.26 (1H, dd, J=17.9 Hz and J=1.4 Hz, H-32’, cis); 7.96 (1H, dd, J=11.5 Hz and J=17.9 Hz, H-31’); 8.53, 9.31, 9.34 (each 1H, s, H-5’, H-10’, H-20’); 13C NMR: 11.3, 11.9, 12.2, 17.5, 19.5, 23.2, 24.0, 29.1, 30.3, 32.9, 33.0, 39.3, 41.8, 48.1, 50.1, 51.8, 93.0, 97.2, 104.1, 106.2, 122.6, 128.3, 129.3, 130.5, 131.6, 135.9, 136.1, 136.2, 137.8, 141.6, 145.1, 149.0, 150.8, 155.3, 160.5, 171.9, 172.1 (C1’, C6’, C9’, C173’, C19’); 196.2 (C131’). Absorption spectra in CH2Cl2, λmax, nm (ε): 413 (85 000); 507 (8 900); 538 (8 000); 609 (7 000); 665 (35 200). Procedure for preparation of conjugates 1–5 The mixture of carboxylic acid (12, 17, or 23, 0.1 mmol), amino containing amide (25, or 26, 0.1 mmol), and DCC (23 mg, 0.11 mmol) in CH2Cl2 (5 mL) was stirred at room tem-perature for 2 h, evaporated to dryness, and the residue was then applied on the top of a silica gel column. The column was initially washed with CHCl3 – acetone – CH3COOH (75:24:1) to remove byproducts, washed with CHCl3 (5 mL), and finally the target product was eluted with CHCl3 – MeOH – 7 M solution of NH3 in MeOH (93:5:2, v/v/v). After evaporation the conjugates were dried in vacuo. 173’[2”-(17β-Hydroxy-3-oxopregn-4-en-21-oylamidoethyl)amido]- pyropheophorbide a (conjugate 1) Conjugate 1 (48 mg, 51 μmol, 51%) was obtained as black powder. HRMS, calculated for [C56H69N6O5]+: 905.5329, found: 905.5327; 1H-NMR: -1.65 (1H, br. s, N-H); 0.25, 0.75 (each 3H, s, H-18 and H-19); 1.58 (3H, t, J = 7.6 Hz, H-82’); 1.79 (3H, d, J = 7.3 Hz, H-181’); 3.16, 3.17, 3.39 (each 3H, s, H-21’, H-71’, H-121’); 4.24, 4.48 (each 1H, m, H-171’ and H-81’); 4.95, 5.19 (each 1H, d, J = 19.7 Hz, H-172’); 5.56 (1H, s, H-4); 6.14 (1H, dd, J = 11.5 Hz and J = 1.4 Hz, H-32’, trans); 6.29 (1H, dd, J = 17.9 Hz and J = 1.4 Hz, H-32’, cis); 6.30 (1H, br. t, J = 5.2 Hz, NH-CO); 6.61 (1H, br. t, J = 5.2 Hz, NH-CO); 7.91 (1H, dd, J = 11.5 Hz and J = 17.9 Hz, H-31’), 8.52, 9.04, 9.26 (each 1H, s, H-5’, H-10’, H-20’); 13C-NMR: 11.1; 11.6; 12.0; 15.1; 16.9; 17.3; 19.2; 20.1; 23.0; 23.3; 29.9; 30.0, 30.4, 31.6; 32.6; 32.9; 33.7; 35.2; 35.5; 37.0; 38.2; 39.6; 40.4; 46.5; 48.0; 48.5; 50.1; 51.6; 52.9; 81.6; 94.0; 97.1; 103.9; 105.8; 122.6; 123.6; 127.8; 129.0; 129.9; 131.7; 135.9; 136.0; 136.3; 137.5; 141.7; 145.1; 148.8; 150.7, 155.4; 160.1, 171.2; 171.8; 173.6; 173.7; 196.3; 199.3. Absorption spectrum is presented in Fig. 2. 173’[5”-(17β-Hydroxy-3-oxopregn-4-en-21-oylamidopentyl)amido]-pyropheophorbide a (conjugate 2) Conjugate 2 (48 mg, 51 μmol, 51%) was obtained as black powder. HRMS, calculated for [C59H75N6O5]+: 947.5793; found: 947.5789; 1H NMR: −1.66 (1H, br. s, N–H); 0.73, 1.00 (each 3H, s, H-18 and H-19); 1.64 (3H, t, J=7.6 Hz, H-82’); 1.78 (3H, d, J=7.3 Hz, H-181’); 3.19, 3.37, 3.46 (each 3H, s, H-21’, H-71’, H-121’); 4.27, 4.46 (each 1H, m, H-171’ and H-81’); 5.00, 5.16 (each 1H, d, J=19.7 Hz, H-151’); 5.28 (1H, br. t, J=5.2 Hz, NH-CO); 5.53 (1H, s, H-4); 6.15 (1H, dd, J=11.5 Hz and J=1.4 Hz, H-32’, trans); 6.25 (1H, dd, J=17.9 Hz and J=1.4 Hz, H-32’, cis); 6.33 (1H, br. t, J=5.2 Hz, NH-CO); 7.94 (1H, dd, J=11.5 Hz and J=17.9 Hz, H-31’); 8.51, 9.28, 9.31 (each 1H, s, H-5’, H-10’, H-20’); 13C NMR: 11.3, 12.0, 12.2, 13.9, 17.3, 17.5, 19.5, 20.6, 23.1, 23.6, 23.9, 28.8, 29.0, 29.8, 30.5, 31.7, 32.7, 33.1, 33.9, 35.7, 36.2, 36.5, 38.6, 39.0, 39.1, 42.6, 46.2, 48.1, 50.1 (x2), 51.7, 53.8, 82.0, 93.1, 97.3, 104.1, 106.0, 122.7, 123.8, 128.2, 129.2, 130.3, 131.8, 136.0, 136.2, 136.4, 137.8, 141.8, 145.2, 149.1, 150.9, 155.5, 160.5, 171.0, 172.0, 172.5, 173.4, 196.3, 199.4. Absorption spectrum is presented in Fig. 2. 173’[2”-(17α-Hydroxy-3-oxopregn-4-en-21-oylamidoethyl)amido]- pyropheophorbide a (conjugate 3) Conjugate 3 (28 mg, 31 μmol, 31%) was obtained as black powder. HRMS, calculated for [C56H69N6O5]+: 905.5329, found: 905.5335; 1H NMR: −1.67 (1H, br. s, N–H); 0.54, 0.88 (each 3H, s, H-18 and H-19); 1.55 (3H, t, J=7.7 Hz, H-82’); 1.75 (3H, d, J=7.3 Hz, H-181’); 3.13, 3.15, 3.35 (each 3H, s, H-21’, H-71’, H-121’); 4.20, 4.45 (each 1H, m, H-171’ and H-81’); 4.92, 5.15 (each 1H, d, J=19.7 Hz, H-151’); 5.53 (1H, s, H-4); 6.11 (1H, dd, J=11.6 Hz and J=1.4 Hz, H-32’, trans); 6.25 (1H, dd, J=17.8 Hz and J=1.4 Hz, H-32’, cis); 6.24 (1H, br. t, J=5.2 Hz, NH-CO); 6.78 (1H, br. t, J=5.2 Hz, NH-CO); 7.88 (1H, dd, J=11.6 Hz and J=17.8 Hz, H-31’); 8.58, 9.10, 9.24 (each 1H, s, H-5’, H-10’, H-20’); 13C NMR: 11.2, 11.6, 12.1, 15.1, 16.9, 17.3, 19.3, 20.1, 23.0, 23.3, 29.9, 30.5, 31.6, 32.6, 33.0, 33.7, 35.2, 35.5, 37.0, 38.2, 39.6, 40.0, 40.5, 46.5, 48.1, 48.5, 50.1, 51.7, 53.0, 81.6, 93.0, 97.1, 103.9, 105.8, 122.7, 123.6, 127.8, 129.0, 129.9, 131.7, 135.9, 136.0, 136.3, 137.6, 141.7, 145.1, 148.9, 150.8, 155.4, 160.2, 171.2, 171.9, 173.6, 173.8, 196.3, 199.3. Absorption spectrum is presented in Fig. 2. 173’[5”-(17α-Hydroxy-3-oxopregn-4-en-21-oylamidopentyl)amido]- pyropheophorbide a (conjugate 4) Conjugate 4 (44 mg, 46 μmol, 46%) was obtained as black powder. HRMS, calculated for [C59H75N6O5]+: 947.5793; found: 947.5793; 1H NMR: −1.67 (1H, br. s, N–H); 0.25, 0.81 (each 3H, s, H-18 and H-19); 1.61 (3H, t, J=7.6 Hz, H-82’), 1.78 (3H, d, J=7.3 Hz, H-181’), 3.17, 3.36, 3.37 (each 3H, s, H-21’, H-71’, H-121’); 4.26, 4.46 (each 1H, m, H-171’ and H-81’); 5.00, 5.15 (each 1H, d, J=19.7 Hz, H-151’); 5.53 (1H, br. t, J=5.2 Hz, NH-CO); 5.58 (1H, s, H-4); 6.14 (1H, dd, J=11.5 Hz and J=1.4 Hz, H-32’, trans); 6.22 (1H, br. t, J=5.2 Hz, NH-CO); 6.23 (1H, dd, J=17.9 Hz and J=1.4 Hz, H-32’, cis); 7.92 (1H, dd, J=11.5 Hz and J=17.9 Hz, H-31’); 8.50, 9.24, 9.32 (each 1H, s, H-5’, H-10’, H-20’); 13C NMR: 11.3, 11.9, 12.1, 15.2, 17.1, 17.4, 19.4, 20.3, 23.1, 23.4, 23.7, 28.5, 28.9, 30.2, 30.6, 31.8, 32.0, 33.1, 33.9, 35.4, 35.7, 37.1, 38.4, 38.9, 39.0, 40.3, 46.6, 48.1, 48.7, 50.1, 51.7, 53.2, 81.8, 93.1, 97.2, 104.0, 105.9, 122.7, 123.7, 128.1, 129.1, 130.2, 131.8, 136.0, 136.2, 136.4, 137.8, 141.7, 145.2, 149.0, 150.8, 155.4, 160.5, 171.5, 172.0, 172.8, 173.1, 196.3, 199.6. Absorption spectrum is presented in Fig. 2. 173’[2’’-(17β-Hydroxy-3-oxopregnan-21-oylamidoethyl)amido]-pyropheophorbide a (conjugate 5) Compound 5 (33 mg, 37 μmol, 69 %) was obtained as black powder. HRMS, calculated for [C56H71N6O5]+: 907.5486, found: 907.5490. 1H NMR: –1.67, (1H, br.s, N–H); 0.56, 0.78 (each 3H, s, H–18 and H–19); 1.65 (3H, t, J=7.6 Hz, 82’); 1.77 (3H, d, J=7.3 Hz, 181’); 3.21, 3.37, 3.45 (each 3H, s, H-21’, H-72’, H-121’); 4.27, 4.46 (each 1H, m, H-171’, H-81’); 5.02, 5.21 (each 1H, d, J=19.7 H-172’); 5.86 (1H, br.t, J=5.2 Hz, NH–CO); 6.14 (1H, dd, J=11.5 Hz and J=1.4 Hz, H-32, cis), 6.20 (1H, dd, J=17.9 Hz and J=1.4 Hz, H-32’, trans), 6.58 (1H, br. t, J=5.2 Hz, NH–CO); 7.93 (1H, dd, J=11.5 Hz and J=17.9 Hz, H-31’); 8.52, 9.30, 9.33 (each 1H, s, H-5’, H-10’, H-20’); 13C NMR: 11.4, 12.0, 13.6, 14.0, 17.3, 19.3, 20.6, 23.1, 23.4, 28.4, 28.7, 29.7, 31.5, 31.9, 33.0, 33.9, 35.3, 35.7, 36.2, 38.5, 39.7, 42.5, 46.0, 46.6, 48.0, 50.0 (x2), 51.7, 53.7, 81.9, 93.1, 7.1, 103.9, 105.9, 122.7, 124.2, 125.3, 128.2, 129.0, 130.0, 131.7, 136.0, 136.3, 137.6, 137.9, 141.7, 145.0, 148.7, 148.9, 160.5, 172.0, 173.6, 174.2, 174.5, 196.3, 211.5; 49. Absorption spectrum is presented in Fig. 2. N(α)-Fmoc-N(ε)-Boc-Lys-hexadecyl amide (28) N(α)-Fmoc-N(ε)-Boc-Lys 27 (200 mg, 0.427 mmol) and DCC (97 mg, 0.47 mmol) were dissolved in dry CH2Cl2 (12 mL), after addition of hexadecyl amine (103 mg, 0.427 mmol), the mixture was stirred for 1 h, diluted with CH2Cl2, washed with NaHCO3 saturated solution (20 mL), water (20 mL), brine (20 mL), dried over Na2SO4 and evaporated to obtain amide 28 (257 mg, 0.371 mmol, 87 %) as white powder. HRMS, calculated for [C42H66N3O5]+: 692.4997, found: 692.4988. 1H NMR: 0.87 (3Н, t, J=6.7 Hz, СH3-hexadecyl), 1.24 (28H, m, (СН2)14-hexadecyl), 1.42 (9Н, s, CH3-Boc), 3.09 (2Н, q , J=5.8 Hz, NСН2(ε)-Lys), 3.21 (2Н, q, J=5.4 Hz, NСН2-hexadecyl), 4.07 (1Н, m, СН(α)-Lys), 4.19 (1H, t, J=6.6 Hz, CH-Fmoc), 4.39 (2Н, d, J=5.2 Hz, СH2-Fmoc), 4.58 (1Н, br.t, NH(ε)-Lys), 5.47 (1Н, br.t, NH- hexadecyl), 6.09 (1Н, br.d, NH(α)-Lys), 7.29 (3Н, t, J=7.4 Hz, Ar-Fmoc), 7.38 (3Н, t, J=7.3 Hz, Ar-Fmoc), 7.57 (2Н, d, J=7.1 Hz, Ar-Fmoc), 7.75 (2Н, d, J=7.4 Hz, Ar-Fmoc). 13C NMR: 14.1, 22.5, 22.7, 24.9, 26.9 (×2), 28.4 (×3), 29.3, 29.4, 29.5, 29.6, 29.7 (×7), 31.9, 32.2, 34.0, 39.6, 47.2, 54.9, 67.0, 78.9, 120.0 (×2), 125.0 (×2), 127.1 (×2), 127.7 (×2), 141.3 (×2), 143.8 (×2), 156.2, 171.5. N(ε)-Boc-Lys-hexadecyl amide (29) The mixture of amide 28 (1.512 g, 2.19 mmol), piperidine (220 μL, 3 mmol) and dry DMF (20 mL) was stirred for 1 h, then poured into ice water (200 mL), stirred for 20 min, the resulted precipitate was filtered, washed with water and dried to obtain N(ε)-Boc-Lys-hexadecyl amide 29 (904 mg, 1.92 mmol, 88 %) as white solid. HRMS, calculated for [C27H56N3O3]+: 470.4316, found: 470.4313. 1H NMR: 0.86 (3Н, t, J=6.7 Hz, СH3-hexadecyl), 1.24 (26H, m, (СН2)13-hexadecyl), 1.42 (9Н, s, CH3-Boc), 3.10 (2Н, q, J=5.31 Hz, NСН2(ε)-Lys), 3.21 (2Н, q, J=6.3 Hz, NСН2-hexadecyl), 3.32 (1H, dd J1=4.3 Hz, J2=7.7 Hz, СН(α)-Lys), 4.17 (1Н, br.t, NH-hexadecyl), 4.56 (1Н, br.t, NH(ε)-Lys); 13C NMR: 14.1, 22.7, 22.9, 24.9, 25.6, 27.0, 28.4 (×3), 29.3, 29.7 (×8), 29.9, 31.9, 34.6, 39.1, 40.2 (×2), 49.1, 55.1, 79.1, 156.1, 174.7. N(α)-173’(Pyropheophorbide)carboxamido-Lys-hexadecyl amide (30) The solution of Pyro (300 mg, 0.56 mmol) and DCC (120 mg, 0.58 mmol) in dry CH2Cl2 (25 mL) was stirred for 30 min, then N(ε)-Boc-Lys-hexadecyl amide 29 (263 mg, 0,56 mmol) was added and the mixture was stirred for 40 min and evaporated to dryness. The residue was separated by silica gel flash chromatography in CH2Cl2 – acetone (9:1) mixture to obtain N(α)-Boc protected conjugate (329 mg, 0,33 mmol, 60 %) as black foam. HRMS, calculated for [C60H88N7O5]+: 986.6841; found: 986.6844. 1H NMR: -1.76 (1H, br.s, NH), 0.86 (3Н, t, J=6.9 Hz, СH3-hexadecyl), 1.18 (28H, m, (CH2)14-hexadecyl), 1.29 (9Н, s, CH3-Boc), 1.66 (3H, t, J=7.6 Hz, H-82’), 1.78 (3H, d, J=7.3 Hz, H-181’), 2.89 (2H, m, NСН2-hexadecyl), 3.09 (2Н, q, J=4.7 Hz, NСН2(ε)-Lys), 3.22, 3.38, 3.58 (each 3H, s, H-21’, H-71’, H-121’), 4.11 (1H, m, H-171’), 4.28 (1Н, m, H-171’), 4.47 (1H, m, H-81’), 4.54 (1Н, br. t, NH(ε)-Lys), 5.04, 5.25 (each 1H, d, J=20.0 Hz, H-151’), 6.00 (1Н, br. d, NH(α)-Lys), 6.15 (1H, dd J1=11.7 Hz, J2=1.4 Hz, H-32’, trans), 6.15 (br.t, 1H, NH-hexadecyl), 6.24 (1H, dd, J1=17.9 Hz, J2=1.4 Hz, H-32’, cis), 7.96 (1H, dd, J1=11.6 Hz, J2=17.8 Hz, H-31’), 8.55, 9.36, 9.42 (each 1H, s, H-5’, H-10’, H-20’). 13C NMR: 11.2, 12.1, 14.1, 17.3, 19.5, 22.4, 22.6, 23.1, 26.8, 28.3 (×3), 29.6 (×14), 30.0, 31.6, 31.9, 32.4, 39.5, 39.8, 48.1, 50.0, 51.7, 52.9, 79.0, 93.3, 97.1, 103.9, 106.3, 122.6, 128.6, 129.1 (×2), 130.6, 131.74, 136.0, 136.3, 137.9, 141.6, 144.8, 149.1, 156.1, 160.6 (×2), 171.4, 171.7, 172.3, 196.1. The product obtained (329 mg, 0.33 mmol), dioxane (10 mL) and 30% aqueous H2SO4 were stirred for 45 min, the removal of Boc-group was controlled by TLC. Thereafter the mixture was poured into the mixture of water (30 mL) and chopped ice (30 g), neutralized with NH4OH, and extracted with dichloromethane (3×25 mL). The combined extract was washed with brine (30 mL), dried over Na2SO4 and evaporated to obtain compound 30 (284 mg, 0.32 mmol, 97 %) as black solid. HRMS, calculated for [C55H80N7O3]+: 886.6317; found: 886.6318. 1H NMR: -1.72 (1H, br.s, NH), 0.86 (3Н, t, J=6.9 Hz, СH3-hexadecyl), 1.19 (28H, m, (CH2)14-hexadecyl), 1.67 (3H, t, J=7.6 Hz, H-82’), 1.77 (3H, d, J=7.3 Hz, H-181’), 3.12 (2Н, q, J=4.67 Hz, NСН2(ε)-Lys), 3.21, 3.38, 3.59 (each 3H, s, H-21’, H-71’, H-121’), 4.20 (1Н, br.t, NH-hexadecyl), 4.28 (1H, m, H-171’), 4.45 (1H, m, H-81’), 5.04, 5.23 (each 1H, d, J=19.9 Hz, H-151’), 5.92 (1Н, br.d, NH(α)-Lys), 6.15 (1H, dd J1=11.5 Hz, J2=1.4 Hz, H-32’, trans), 6.26 (1H, dd, J1=18.0 Hz, J2=1.4 Hz, H-32’, cis), 7.97 (1H, dd, J1=11.7 Hz, J2=17.8 Hz, H-31’), 8.52, 9.34, 9.41 (each 1H, s, H-5’, H-10’, H-20’). 13C NMR: 11.3, 12.1, 12.2, 14.2, 17.5, 19.5, 22.6, 22.8, 23.2, 23.8, 27.0 (×10), 32.0, 32.6, 32.8, 39.5, 39.6, 41.5, 48.1, 50.1, 50.9, 51.7, 53.2, 93.0, 97.2, 104.1, 106.2, 122.6, 128.2, 129.3 (×2), 131.6, 135.9, 136.1, 137.7, 137.9, 141.5, 141.6, 145.1, 148.9, 150.9, 155.4, 160.4, 171.6, 172.1, 196.2. N(α)”-173’(Pyropheophorbide)carboxamido-N(ε)”-21(17β-hydroxy-3-oxo- pregn-4-ene-21-oyl)amido-Lys-hexadecyl amide (conjugate 6) DCC (100 mg, 0.49 mmol) was added to the stirred solution of 17β-hydroxy-3-oxopregn-4-en-21-oic acid 12 in dry CH2Cl2 (15 mL); the mixture was stirred for 10 min; then compound 30 (217 mg, 0.25 mmol) was added, and the mixture was stirred for 12 h more. Thereafter the mixture was evaporated, the residue was separated by silica gel flash chromatography in CH2Cl2 – acetone – AcOH (84:15:1), evaporated, and dried to obtain conjugate 6 as black amorphous powder (141 mg, 0.12 mmol, 47 %). HRMS, calculated for [C76H108N7O6]+: 1214.8356; found: 1214.8363. 1H NMR: -1.67 (1H, br.s, NH), 0.71 (3H, s, H-18), 0.86 (3Н, t, J=7.0 Hz, СH3-hexadecyl), 0.95 (3H, s, Н-19), 1.17 (28H, m, (CH2)14-hexadecyl), 1.60 (3H, t, J=7.6 Hz, H-82’), 1.74 (3H, d, J=7.2 Hz, H-181’), 2.34 (2Н, AB system, Н-20’), 3.11 (2Н, q, J=6.5 Hz, NСН2(ε)-Lys), 3.14, 3.33, 3.40 (each 3H, s, H-21’, H-71’, H-121’), 4.24 (1H, m, H-171’), 4.39 (1H, m, H-81’), 4.88, 5.13 (each 1H, d J=20.0 Hz, H-151’), 5.28 (1H, s, 17-OH), 5.48 (1H, s, Н-4), 6.12 (1H, dd, J1=11.6 Hz, J2=1.4 Hz, H-32’, trans), 6.22 (1H, dd, J1=17.9 Hz, J2=1.4 Hz, H-32’, cis), 6.40 (1H, br.d, NH(α)-Lys), 6.46 (1Н, br. t, NH-hexadecyl), 6.72 (1Н, br.t, NH(ε)-Lys), 7.88 (1H, dd, J1=14.7 Hz, J2=17.8 Hz, H-31’), 8.46, 9.19, 9.23 (each 1H, s, H-5’, H-10’, H-20’). 13C NMR: 11.1, 11.8, 12.0, 13.7, 14.1, 17.1, 17.6, 19.3, 20.4, 22.6, 22.7, 22.9, 23.4, 26.9, 28.6, 29.2, 29.6 (×10), 30.4, 31.4, 31.5, 31.8, 31.9, 32.5, 32.8, 33.8, 35.5, 36.1, 36.1, 38.4, 38.6, 39.6, 42.5, 46.0, 48.0, 49.8, 50.0, 51.5, 52.9, 53.5, 81.9, 92.9, 97.1, 103.9, 105.7, 122.6, 123.7, 128.2, 129.1, 130.0, 131.6, 135.9, 136.0, 136.3, 137.6, 141.7, 145.0, 148.9, 150.8, 155.4, 160.2, 170.9, 171.5, 171.7, 172.7, 173.4, 196.3, 199.2. N(α)-Fmoc-Lys-hexadecyl amide (31) The mixture of amide 28 (911 mg, 1.32 mmol), CH2Cl2 (30 mL), and TFA (10 mL) was stirred for 30 min, evaporated to dryness, the residue was dissolved in CH2Cl2 (30 mL), the solution was washed with saturated NaHCO3 solution (20 mL), brine (20 mL), dried over Na2SO4, and evaporated to obtain N(α)-Fmoc-Lys-hexadecyl amide 31 (766 mg, 1.29 mmol, 98 %) as black film. HRMS, calculated for [C37H58N3O3]+: 592.4473; found: 592.4477. 1H NMR: 0.87 (3Н, t, J=6.3 Hz, СH3-hexadecyl), 1.36 (28H, m, (CH2)14), 2.69 (2H, m, CH2(ε)-Lys), 3.22 (2Н, m, NСН2-hexadecyl), 4.07 (1Н, m, СН(α)-Lys), 4.20 (1H, t, J=6.6 Hz, CH-Fmoc), 4.40 (2Н, d, J=5.9 Hz, СH2-Fmoc), 5.50 (1Н, br.t, NH-hexadecyl), 6.16 (1Н, br. d, NН(α)-Lys), 7.30 (3Н, t, J=7.4 Hz, Ar-Fmoc), 7.39 (3Н, t, J=7.4 Hz, Ar-Fmoc), 7.57 (2Н, d, J=7.4 Hz, Ar-Fmoc), 7.75 (2Н, d, J=7.6 Hz, Ar-Fmoc). 13C NMR: 14.2, 22.7, 22.8, 27.0, 29.4, 29.4, 29.8 (×11), 32.0, 32.6, 39.7, 41.7, 47.3, 55.1, 67.1, 120.1 (×2), 125.1 (×2), 127.2 (×2), 127.8 (×2), 141.4 (×2), 143.9 (×2), 171.5. N(ε)-173’(Pyropheophorbide)carboxamido-Lys-hexadecyl amide (32) The solution of Pyro (250 mg, 0,47 mmol) and DCC (97 mg, 0.47 mmol) in dry CH2Cl2 (25 mL) was stirred for 30 min, after addition of N(α)-Fmoc-Lys-hexadecyl amide 31 (227 mg, 470 μmol) the mixture was stirred for 45 min, and evaporated. TLC analysis revealed partial deletion of Fmoc-group in the resultant product. The residue dissolved in DMF (5 mL) was mixed with piperidine (37 μL, 0.5 mmol). The resultant mixture was stirred for 1 h, diluted with CH2Cl2 (30 mL), washed with water (2×10 mL), dried over Na2SO4, and evaporated. The residue was separated by silica gel flash chromatography in CH2Cl2 – acetone (93:7) mixture to obtain compound 32 (146 mg, 170 μmol, 35 %). HRMS, calculated for [C55H80N7O3]+: 886.6317; found: 886.6307. 1H NMR: -1.69 (1H, br.s, NH), 0.86 (3Н, t, J=7.5 Hz, СH3-hexadecyl), 1.23 (28H, m, (CH2)14), 1.65 (3H, t, J=7.6 Hz, H-82’), 1.78 (3H, d, J=7.3 Hz, H-181’), 2.98 (2Н, q, J=6.2 Hz, NСН2(ε)-Lys), 3.20, 3.38, 3.51 (each 3H, s, H-21’, H-71’, H-121’), 4.21 (1H, m, H-171’), 4.50 (1H, m, H-81’), 5.05, 5.23 (each, 1H, d, J=19.9 Hz, H-151’), 5.42 (1H, br.t, NH-hexadecyl), 6.15 (1H, dd, J1=11.6 Hz, J2=1.4 Hz, H-32’, trans), 6.27 (1H, dd, J1=17.8 Hz, J2=1.4 Hz, H-32’, cis), 7.14 (1H, br.t, NH(ε)-Lys), 7.97 (1H, dd, J1=11.5 Hz, J2=17.8 Hz, H-31’), 8.52, 9.34, 9.35 (each 1H, s, H-5’, H-10’, H-20’). 13C NMR: 11.3, 12.0, 12.2, 14.2, 17.5, 19.5, 22.8, 23.1, 23.2, 23.9, 27.0, 28.9, 29.0, 29.8 (×8), 30.4, 30.5, 32.0, 33.0, 34.3, 38.9, 39.1, 48.2, 50.1, 51.9, 54.8, 93.1, 97.2, 104.1, 106.2, 122.6, 128.3, 128.9, 129.3, 130.9, 131.7, 136.0, 136.1, 136.3, 141.6, 145.1, 149.1, 150.8, 155.3, 160.6, 172.0, 172.4, 174.5, 196.4. N(α)”-21(17β-Hydroxy-3-oxopregn-4-ene-21-oyl)amido-N(ε)”-173’(pyropheophorbide)carboxamido-Lys-hexadecyl amide (conjugate 7) The mixture of compounds 32 (65 mg, 73 μmol), 12 (26 mg, 73 μmol), and DCC (17 mg, 80 μmol) was stirred for 25 min, the reaction was controlled by TLC. After mixture evaporated, the residue was applied on the top of silica gel column, the column was washed with CHCl3 –acetone – AcOH (85:14:1) and the target product was then eluted with CHCl3 – acetone – AcOH (79:20:1). The isolated crude conjugate was additionally purified by silica gel flash chromatography in CHCl3 – MeOH – AcOH (93:6:1) to obtain conjugate 7 (47 mg, 39 μmol, 53 %) as black powder. HRMS, calculated for [C76H108N7O6]+: 1214.8356; found: 1214.8362. 1H NMR: -1.66 (1H, br.s, NH), 0.70 (3H, s, H-18), 0.86 (3Н, t, J=7.0 Hz, СH3-hexadecyl), 0.91 (3H, s, Н-19), 1.21 (28H, m, (CH2)14), 1.61 (3H, t, J=7.6 Hz, H-82’), 1.76 (3H, d, J=7.1 Hz, H-181’), 2.29 (2Н, AB system, Н-20), 3.11 (2Н, q, J=6.5 Hz, NСН2(ε)-Lys), 3.16, 3.34, 3.38 (each 3H, s, H-21’, H-71’, H-121’), 4.30 (1H, m, H-171’), 4.44 (1H, m, H-81’), 4.97, 5.14 (each 1H, d, J=19.9 Hz, H-151’), 5.49 (1H, s, Н-4), 5.80 (1H, br.t, NH-hexadecyl), 6.12 (1H, dd, J1=11,5 Hz, J2=1.4 Hz, H-32’, trans), 6.22 (1H, dd, J1=18.0 Hz, J2=1.4 Hz, H-32’, cis), 6.65 (1H, br. t, NH(ε)-Lys), 7.15 (1Н, br.d, NH(α)-Lys), 7.89 (1H, dd, J1=14.7 Hz, J2=17.9 Hz, H-31’), 8.47, 9.22, 9.25 (each 1H, s, H-5’, H-10’, H-20’). 13C NMR: 11.3, 11.9, 12.1, 13.9, 14.2, 17.1, 17.4, 19.4, 20.4, 22.8, 23.1, 23.5, 27.0, 29.0, 29.4, 29.4, 29.5, 29.6, 29.8 (×10), 30.8, 31.3, 31.5, 31.6, 32.0, 32.6, 33.4, 33.8, 35.5, 35.7, 36.2, 38.4, 38.8, 39.7, 42.9, 46.3, 48.1, 49.9, 50.1, 51.8, 53.1, 53.4, 81.1, 93.8, 97.2, 104.1, 105.7, 122.7, 123.8, 128.0, 129.1, 130.0, 131.7, 136.0, 136.2, 136.4, 137.7, 141.8, 145.1, 149.1, 150.9, 155.5, 160.5, 170.8, 171.6, 172.0, 172.8, 173.4, 196.4, 199.2. Molecular modeling Conformation searches have been performed using molecular mechanics MMFF94 force field parameters in vacuo. OpenBabel package [26] was employed for initial structure preparation and energy minimizations. Simulated annealing molecular dynamics (MD) has been performed to sample low-energy conformation space of conjugates by means of NAMD [27] software. Parameters and topology files were generated with the aid of the SwissParam server [28] on the basis of the MMFF94 force field. The annealing protocol consisted of 4 ps high temperature runs at 500 K followed by 4 ps cooling phase bringing temperature down to 50 K, with total of 200 annealing cycles scheduled in 32 processes. This procedure yielded 6400 local energy minima for each compound. Resulting structures were then optimized by energy minimization with MMFF94 potential. VMD package [29] was used for MD trajectory post-processing, analysis, and visualization. Solubilization of conjugates 6 and 7 in aqueous medium Solubilization of conjugates with PC To obtain solution conjugate/PC at ratio of 1:10 (mg/mg, 6.7 molar % of conjugate) calculated volumes of 10-2 M solutions of PC and conjugate (either 6, or 7) in CHCl3 were mixed together. Mixed solutions were evaporated to dryness, and dissolved in i-PrOH at 40°C to obtain solutions with concentrations of conjugates equal to 10-3 M. Aliquots of heated isopropanolic solutions were injected during vortexing into 100-fold volume of PBS (for measuring of absorption spectra and particle size distributions) or in culture medium (for measuring of uptake and internalization of conjugates by cells). Solubilization of conjugates with Pluronic F68 Calculated volumes of 10-2 M solutions of pluronic F68 and conjugates (either 6, or 7) in CHCl3 at ratios 1:10 and 1:50 (mg/mg) were mixed together to obtain solutions conjugate/pluronic. Mixed solutions were evaporated to dryness, then calculated volumes of PBS, or culture medium were added to films, and the mixtures obtained were vortexed at 40 °C for 1 min. Biological evaluation Cell cultures The human prostate carcinoma LNCaP and PC-3 cells, breast carcinoma MCF-7 cells, hepatocarcinoma Hep G2 cells were obtained from the American Type Culture Collection (USA). Cells were propagated in culture dishes at the desired densities in RPMI 1640 and DMEM medium supplemented with 10% fetal calf serum (FCS; Gibco, USA) and 1% penicillin/streptomycin “Gibco” in a 5% CO2 atmosphere at 37 °C for 24 h. Before experiments the cells were seeded either in 96-well plates at a density of 5·103 cells/well (for MTT assay), or in 6-well plates at a density of 106 cells/well (for investigation of uptake and internalization of conjugates) and incubated for 48 h. Uptake and internalization of conjugates by prostate carcinoma cells LNCaP cells seeded in a 6-well plates were incubated for 2 h, 6 h, 14 h and 20 h at 37 °C with conjugates 1–4 (25 μM in culture medium), then medium was aspirated, cells were washed with cold PBS at 4 °C, and lipids were extracted from each well with hexane – i-PrOH mixture (3:2, 3×0.5 mL). Pellets were used for cell protein concentration measurements [30]. The lipid extracts were dried under nitrogen flow, residues were dissolved in CH2Cl2 (2 mL), and concentrations of conjugates were determined spectrophotometrically by means of a “Cary Spectra 100” spectro-photometer in CHCl3 and CH2Cl2 using a quartz cell with the 1 mm optical path length. All measurements were carried out in triplicates. The efficiency of cell labeling was expressed in terms of ratios of internalized conjugates (nmol per mg of cell protein). MTT cell viability assay LNCaP and PC-3 cells were treated with conjugates at the concentrations of 0.1 µM, 1 µM, 5 µM, 10 µM, 25 µM, 50 µM, 100 µM and incubated for 96 h in 96-well plates. Then, solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/mL) was added and the cells were incubated for 4 h, followed by measuring absorbance at 570 nm, with a “Techan Genius plus” microplate reader. The viability of treated cells was expressed as a percentage relative to that of control cells. Each experiment was performed in triplicate, and independently repeated at least four times. Measuring of photo induced toxicity LNCaP cells were incubated with concentrations of conjugates 1 and 5 at ranged from 0.1 μM to 100 μM for 18 h in RPMI 1640 and DMEM medium supplemented with 10% fetal calf serum (FCS; Gibco, USA) and 1% penicillin/streptomycin “Gibco” in a 5% CO2 atmosphere, then the cells were washed three times with PBS, thereafter fresh culture medium was added (100 μL/well), and cells were irradiated for 10 min with light (λ = 660 nm) using a LED AFS "Spectrum" instrument (р = 0,142 W; 85,2 J/cm2). Then the cells were incubated in fresh medium for 24 h at 37 °C. The viability of irradiated cells and control cells (incubated similarly except irradiation) was measured using MTT assay. All experiments were carried out in triplicates. RESULTS AND DISCUSSION Chemical synthesis Preparation of new conjugates 1 – 7 (consisted of synthesis steroidal blocks 12, 17 and 23; synthesis of bifuntional conjugates 1 – 5 by coupling of Pyro with steroidal blocks 12, 17 and 23 by means of diamino containing linkers; and synthesis of trifunctional conjugates 6 and 7) is presented in the Schemes 1, 2, and 3, respectively. Synthesis of steroidal acids 12, 17, 23 was performed as follows. Testosterone 8 and dihydrotestosterone 13 were transformed to steroid blocks 12 and 17 by five steps including consecutive protection of carbonyl functions with formation of 1,3-dioxolanes [13], oxidation of 17β-hydroxyl groups [14], and Reformatsky reaction of obtained 17-ketones 10 and 15 with Zn and ethyl bromoacetate [15, 16]. The aforementioned reaction is known to pass stereoselectively and give an appropriate 17β-OH isomer. The consecutive removal of ethylene ketal and ethyl ester protective groups led to 21-carboxylic acids 12 and 17 in 49 % and 58 % overall yields (based on compounds 8 and 13, respectively). The steroidal block 23 was synthesized from cyclosteroid 18 [17] in five steps. The introdution of 17α-hydroxyl group was carried out by oxidation of 17(20)-double bond with m-chloroperbenzoic acid followed by the reduction of resulting of 17α,20-epoxide 19 [the mixture of related 17α,20(R)- and 17α,20(S)- isomers in the ratio of 3: 1] with LiAlH4 in boiling THP to obtain 17α,21-diol 20. Then diol 20 was transformed to hydroxy acid 21 by oxidation with ruthenate – potassium bromate reagent [18] in acetone – water (3:1) solution. Then acid 21 was subjected to acid hydrolysis to obtain 3β-hydroxy-5-ene acid 22. Oxidation of compound 22 with Dess-Martin periodinane, followed by acid catalyzed isomerization of crude 3-oxo-5-ene resulted in target 17α-hydroxy-3-oxopregn-4-en-21-oic acid 23 in 22 % overall yield (based on compound 18). Both 17β-hydroxy- and 17α-hydroxy acids 12, 17 and 23 were obtained as pure compounds; the configuration of C17 was confirmed by 13C NMR spectra. The differences in chemical shifts of C16, C17, and C18 resonances in compounds 12, 17 and 23 were consistent with published data of 13C NMR spectra for related 17α- and 17β-hydroxyestradiols [19].
Synthesis of bifunctional conjugates 1–5 is presented in Scheme 2. Initially, Pyro was transformed to related pentafluorophenyl ester 24, which was then treated with excess of either ethylene diamine or 1,5-diaminopentane to obtain amides 25 and 26 comprising primary amino group. Condensation of compounds 25 and 26 with steroidal acids 12, 17 and 23 in the presence of DCC led to the target conjugates 1–5.
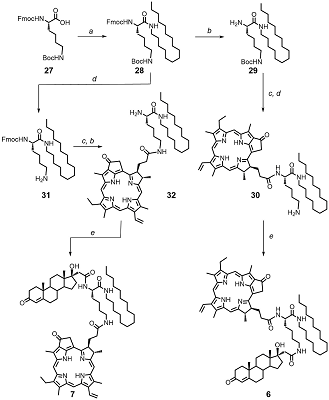
Synthesis of trifunctional conjugates 6 and 7 is presented in Scheme 3. Initially, N(α)-Fmoc-N(ε)-Boc-Lys 27 was condensed with hexadecyl amine to obtain protected lysyl amide 28. To prepare conjugate 6, amide 28 was consequently treated with piperidine to remove the Fmoc-protecting group; then the amino containing block 29 was coupled with Pyro. To remove Boc-protecting group the obtained intermediate was treated in acidic conditions; and finally the resulting amine 30 was acylated with steroidal acid 12 to obtain conjugate 6. We observed sufficient racemization of C17 in 17-hydroxy-3-oxopregn-4-en-21-oyl amides in the conditions of the group removal. For this reason we changed the consequence of reactions as follows: initially we removed Boc-protective group in amide 28 and coupled the obtained amine 31 with Pyro (wherein the partial removal of Fmoc-protecting group was observed); then, after complete Fmoc-group deletion, the amine 32 was condensed with steroidal acid 12 to obtain target conjugate 7.
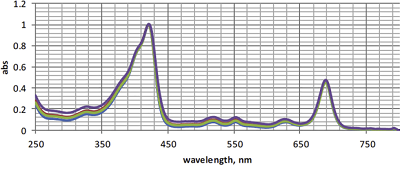
All synthesized conjugates 1-7 were prepared as pure compounds, their structures were completely characterized by HRMS, 1H NMR, 13C NMR and absorption spectra. Spectral properties and molecular models The absorption spectra of conjugates 1–7 were similar to each other and were typical for Pyro and their derivatives. The normalized absorption spectra of conjugates 1–4 are presented in the Figure 2.
The 1H NMR spectra of conjugates 1–7 indicated a significant mutual influence of steroid and macrocyclic fragments. The chemical shifts of selected protons in 1H NMR spectra of conjugates 1–4, 6 and 7 (comprising either testosterone or epitestosterone) are shown in Table 1. The strong high-field shifts for H-4, H-18, H-19 resonances were observed in the spectra of conjugates 1–4 in comparison with those in spectra of unconjugated steroidal acids 12 and 23. The highest field shift for H-4 resonance was observed in spectra of conjugates 2 and 3, while that for H-18 resonance – in spectra of conjugates 1 and 4. The modest high field shifts for H-18 and H-19 resonances were observed in spectra of trifunctional conjugates 6 and 7. Chemical shifts for amide NH” resonances strongly depended on the conjugate structure, while those for H-5’, H-10’ and H-20’ resonances in pyropheophorbide a moieties differed insignificantly.
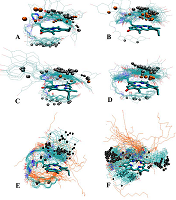
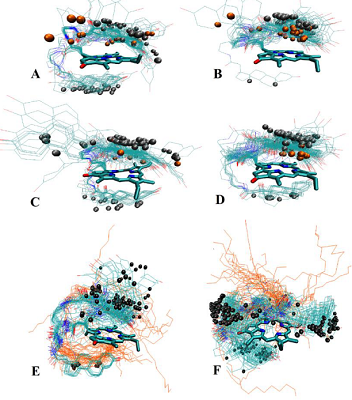
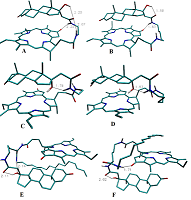
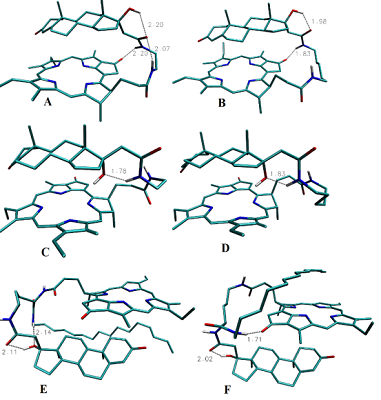
The data presented in the Table 1 are in agreement with results of molecular modeling (performed by simulated annealing) indicated differences in positional relationships of steroid and macrocycle moieties in the conjugates. The calculated ensembles of conformers, truncated at 10 kcal/mol above the lowest-energy conformer, are shown in the Figure 3; the structures for lowest energy conformers are presented in Figure 4.
Configuration of C17 affects the relative positioning of 17-hydroxy group, and in epitestosterone conjugates 3 and 4 this group was found to be oriented towards the macrocycle (Fig. 3C and 3D), and is capable of hydrogen bond formation with nearby amide proton (Fig. 4C and 4D). On the contrary, in testosterone derivatives 1 and 2 this hydroxyl group is directed outwards from the macrocycle, and completely exposed to the environment (Fig. 3A, 3B, 4A and 4B). Due to the longer linker lengths in conjugates 2 and 4, they exhibit higher conformational flexibility in comparison with conjugates 1 and 3. This allows more conformers with NH proton to be axially positioned off the macrocycle plane, pushing corresponding NMR resonances into the higher field. Because position of steroid moiety in the lowest-energy conformers was found to be not significantly different between conjugates with different linker lengths, we have concluded that the main effect of linker lengthening is, indeed, enhanced conformational flexibility. Structures with the steroid moiety hoisted over the surface of macrocycle were found energetically favorable for conjugates 1–4 (Fig. 3A – 3D). This ‘folded’ structure correlates well with observed high-field shifts of 18- and 19-methyl protons as compared to unconjugated steroids, because of shielding effect exerted by a large aromatic moiety of Pyro on atoms located above and below its surface. The presence of ‘unfolded’ conformers with relatively low energy in compounds 2 and 3 (Fig. 3B and 3C) is probably responsible for the observed weaker shielding of 18- and 19-methyls. Figure 3E demonstrates that structures with steroid moiety hoisted over the surface of macrocycle, and hexadecyl chain located in its opposite side, are energetically favorable for trifunctional conjugate 6. On the contrary, Figure 3F reveals that three ensembles of low energy conformers differing in positions of steroid relatively to macrocycle, and random distribution of hexadecyl chain, are favored for trifunctional conjugate 7. In both conjugates 6 and 7 18- and 19-methyl groups were mainly turned away from macrocycle.
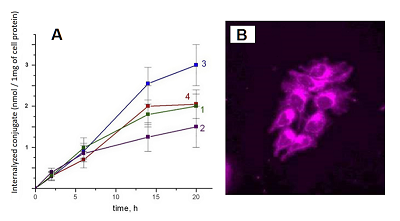
The lowest energy conformers of conjugates are stabilized by possibility of intramolecular hydrogen bonds formation (Fig. 4). In the lowest energy conformers of conjugates 1 and 2 Nα’’ atom is located near carbonyl group of Pyro, thus making formation of corresponding hydrogen bond favorable. Additionally, in compound 1 the oxygen atom of the 21-amide group is located between 17β-hydroxyl group and Nω’’ atom of ethylene diamine linker, and thus can serve as proton acceptor with either of these atoms being a donor (Fig. 4A, 4B). In the lowest energy conformers of conjugates 3 and 4 17α-hydroxyl group is located close to Nα’’ atom, and apparently may serve as proton acceptor to form the corresponding hydrogen bond (Fig. 4C, 4D). In the lowest energy conformers of both conjugates 6 and 7, the hydrogen atom of the steroid 17-hydroxyl group participates in hydrogen bond formation with oxygen of related 21-carboxamido group. Additionally, in the lowest energy conformer of conjugate 6, the oxygen atom of the 17-hydroxyl group is located near the nitrogen atom of hexadecyl amide, and thus may serve as a proton acceptor to form the corresponding hydrogen bond (Figure 4E). In the lowest energy conformer of conjugate 7, the nitrogen atom of hexadecyl amide is located near the carbonyl group of pyropheophorbide a, and may be involved in formation of the corresponding hydrogen bond (Fig. 4F). Interaction of bifunctional conjugates with cultured cells Conjugates 1–5 were efficiently internalyzed by prostate carcinoma LNCaP and PC-3 cells, breast carcinoma MCF-7 cells and hepatocarcinoma Hep G2 cells. The internalisation was dependent on the structure of conjugates. Time course of conjugates 1–4 uptake by LNCaP cells is given in the Fig. 5A. Epitestosterone derivatives were internalyzed more efficiently than testosterone ones; in both pairs conjugates comprising shorter linkers were internalyzed more efficiently than those comprising long linkers (3 > 4 ≥ 1 > 2). According to our molecular models, this dependence must be correlated to diminished conformational flexibility of compounds 1 and 3, combined with predominant 17-hydroxyl group exposure in testosterone derivatives. The conjugates internalization by cells was confirmed as follows: LNCaP cells, initially labeled with conjugates 1–4 for 6 h, were incubated for 12 h in fresh medium, followed by determination of conjugates content in it as described in methods. The absence of detectable amounts of conjugates in the medium proves that they were completely internalized by cells. The photographs of MCF-7 cells labeled with conjugate 1 is presented in the Fig. 5B.
Conjugates 1–5 moderately inhibited the growth and proliferation of LNCaP and PC-3 cells at 24 h incubation, however potently inhibited it at prolong of incubation. The MTT test [20] data demonstrating effects of conjugates 1–5 on the growth and viability of LNCaP and PC-3 cells at 96 h incubation are presented in the Table 2. Conjugates 1–5 inhibited growth of both LNCaP and PC-3 cells, however, effect on LNCaP cells was more pronounced. Anti-proliferative activity of conjugates in LNCaP and PC-3 cells was dependent on the structure of steroid moiety and length of the linker and decreased in the following row: 3 > 1 > 4 > 2 > 5.
Photo induced toxicity of conjugates 1 and 5 in LNCaP and PC-3 cells was also primary evaluated. For this purpose LNCaP and PC-3 cells were incubated with conjugates 1 and 5 for 18 h at concentration of of 0.1 µM, 1 µM, 5 µM, 10 µM, 25 µM, 50 µM, 100 µM , then the labeled cells were irradiated with light (l = 660 nm) for 10 min, thereafter the irradiated cells were incubated for 24 h in fresh medium. The irradiated cells, as well as the control cells (treated by the same way, but without irradiation) were analyzed using the MTT test. The IC50 values for irradiated and non irradiated cells are presented in the Table 3. These data revealed that conjugates 1 and 5 exhibited significant photo induced toxicity in prostate carcinoma cells.
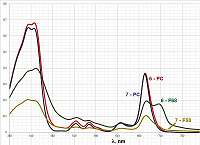
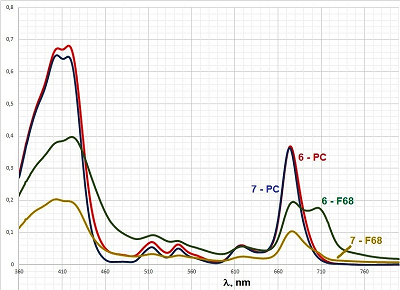
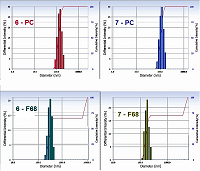
In contrast to bifunctional conjugates 1–5, trifunctional conjugates 6 and 7 were insoluble in aqueous media, so we did not conduct experiments, as described above, with these conjugates. Solubilization of trifunctional conjugates in aqueous medium In order to solubilize trifunctional conjugates 6 and 7 in aqueous media we used two reported earler methods: (i) injection of mixed iso-propanolic solution of conjugate and phosphatidyl choline (PC) into aqueous buffer [12]; (ii) hydration of mixed films conjugate – pluronic F68 [21]. We have prepared mixed micelles 6 – PC and 7 – PC with mass ratio conjugate/PC equal to 1:10 (which corresponded to concentration of 6.7 molar % of conjugates); and micelles 6 – F68 and 7 – F68 with mass ratio conjugate/pluronic equal to 1:10 and 1:50. Absorption spectra and particle size distribution (measured by laser scattering) for these preparations are presented in Figures 6 and 7, respectively.
The spectra of micelles 6 – PC and 7 – PC were nearly identical and highly resolved; the Soret bands had two maxima at 402 nm and 417 nm (the last one is known to be characteristic for the aggregated form of conjugates); the long wave maxima had red shifts of about 6 nm (as compared to those for spectra of conjugates 6 and 7 in CH2Cl2) and were observed at 674 nm; the maxima at 516 nm, 544 nm, 618 nm were clearly visible (Fig. 6). The mean sizes of micelles 6–PC and 7–PC were 123.3 nm and 108.0 nm, respectively (Fig. 7). These mixed micelles possessed high stability – their absorption spectra and particle size distribution did not show any visible changes during the storage for 1 week.
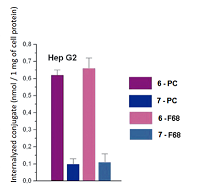
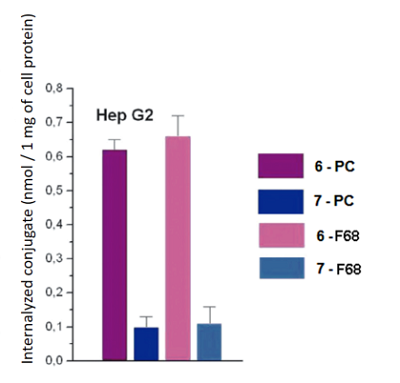
The spectra of micelles 6–F68 and 7–F68 (at the conjugate/pluronic ratio of 1:50) were insufficiently resolved; the Soret bands were broad; the long wave maxima had additional shoulder near 710 mn, that indicated association of macrocycle chromophores with formation of stacked structures [22, 23]. The spectra of micelles 6–F68 and 7–F68 (at the conjugate/pluronic ratio of 1:10) were similar to those presented in Figure 6, but displayed certain turbidity and poor resolution. The mean sizes of conjugate-pluronic micelles 6–F68 and 7– F68 (with the mass ratio conjugate/pluronic 1:50) were 621.3 nm and 385.7 nm, respectively (Fig. 7). The absorption spectra of these micelles displayed significant changes after 24 h of storage, and after 1 week of storage the presence of mixed micelles was undetectable, that indicates low stability of mixed micelles conjugate-pluronic. Mixed micelles of conjugates 6–PC, 7–PC, 6–F68, and 7–F68 poorly interacted with prostate carcinoma LNCaP and PC-3 cells, however significantly internalized by hepatocarcinoma Hep G2 cells. The internalization of conjugate 6 was about 5-fold stronger than that of conjugate 7. It depended on the structure of conjugate, rather than on the method of solubilization – the micelles conjugate – PC and conjugate – pluronic were internalized similarly (Fig. 8).
Apparently, binding of conjugates to Hep G2 cells may be explained by the existence of lipid binding sites (LBS) on the surface of these cells and affinity of hexadecyl moiety of conjugates to LBS [24]. COMPLIANCE WITH ETHICAL STANDARDS This work does not contain any studies using humans and animals as research subjects. FUNDING The work was performed carried out within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021-2030) (№ 122030100170-5). CONFLICTS OF INTEREST The authors declare no conflict of interest. REFERENCES
|