|
Identification of Ribosomal Footprints on the Electrophoretic Gel in Translatome Profiling: on the Use of DNA Size Standards Institute of Biomedical Chemistry, 10 Pogodinskaya str., Moscow, 119121 Russia; e-mail: khmelevaswetlana@yandex.ru Key words: translatome profiling, ribosome footprints, isolation, DNA size standards DOI: 10.18097/BMCRM00195 INTRODUCTION
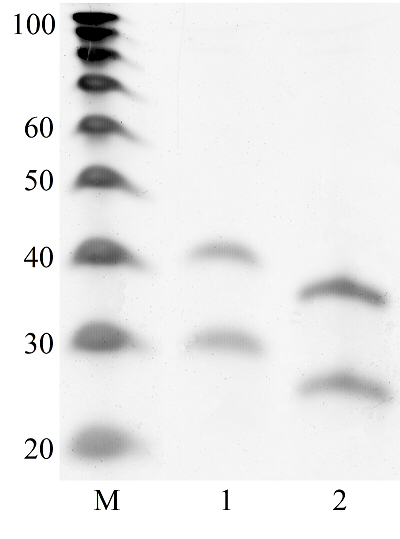
Transcriptome-wide measurement of translation by high-throughput RNA sequencing, known as translatome profiling, has been attracting substantial attention, especially after the work of Ingolia et al. [1] where the approach based on sequencing of ribosome footprints (small fragments of mRNA of about 30 nucleotides (nt) long, protected by ribosome subunits from digestion by an exogenous ribonuclease) has been realized. This approach referred to as ‘Ribo-Seq’ or ‘ribosome profiling’ allows one to quantify the number of mRNA engaged with ribosomes and, thus, likely undergoing a translation and also to learn about the ribosome movement along a mRNA chain and how it might be influenced by mRNA sequence [2]. The common protocol of ribosome profiling, prior to a preparation of sequencing library and RNA sequencing per se, includes steps such as a treatment of a cell or tissue lysate with RNase I, isolation of ribosomes with the protected mRNA fragments, followed by the additional purification of ribosome footprints by denaturing polyacrylamide gel electrophoresis. The last step in turn includes the excision of gel pieces containing ribosome footprints and their subsequent elution out of the gel to further prepare a sequencing library [3]. To select the area of gel containing ribosome footprints, RNA oligonucleotides as size standards are commonly employed [1, 3]. However, RNA is quite demanding to conditions of storage and handling due to its high susceptibility to enzymatic degradation by ubiquitous ribonucleases. Moreover, RNA size standards are expensive since chemical synthesis of RNA oligonucleotides is at least 10 times more costly than that of DNA oligonucleotides. The substitution of RNA oligonucleotides with DNA oligonucleotides as size standards to locate the ribosome footprint band on a polyacrylamide gel would be undoubtedly beneficial. Although the flexibility of single-stranded DNA and RNA linear molecules does not appreciably differ [4, 5], it still can affect the electrophoretic migration of short polynucleotide chains thus precluding a direct substitution of RNA size standards with those of DNA of the same length. The aim of this work was to investigate at how the positions of 25 and 35 nt long RNA oligonucleotides on a denaturing polyacrylamide gel are related to those of DNA oligonucleotides of the same length and nucleotide composition (except for the substitution of U with dT) and whether some commercial DNA size standards can replace the RNA oligonucleotides. MATERIALS AND METHODS The chemicals used were from “Merck” (USA), unless stated otherwise. Their quality was of the ACS grade or higher. Turbo DNase, RNase I, SUPERase·In were purchased from “Thermo Fisher Scientific” (USA, Cat. Nos. AM2239, EN0601, and AM2694, respectively), 20/100 DNA oligo length standards – from “Integrated DNA Technologies” (USA, Cat. No. 51-05-15-02). Solutions and buffers were prepared with nuclease-free water. 25 and 35 nt long RNA (AUGCAUGCAUGCAUGCAUGCAUGCA and AUGCAUGCAUGCAUGCAUGCAUGCAUGCAUGCAUG, respectively) and DNA (ATGCATGCATGCATGCATGCATGCA and ATGCATGCATGCATGCATGCATGCATGCATGCATG, respectively) oligonucleotides were synthesized and purified by “Syntol” (Russia). HepG2 cells (ATCC HB-8065, “ATCC”, USA) were cultured as described in [6]. Prior to harvesting, cells were incubated for 1 min in the presence of cycloheximide added to the culture medium at the final concentration of 0.1 mg/ml. The harvested cells washed 3 times with phosphate buffered saline supplemented with cycloheximide (0.1 mg/ml) were pelleted by centrifugation and lysed in the buffer contaning 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.1 mg/ml cycloheximide, 1% Tritone-100, 25 U/ml Turbo DNase for 10 min on ice. The treatment with RNase I and the isolation of ribosomes with MicroSpin S-400 columns (“Cytiva”, USA, Cat. No. 27514001) were carried out as described in [7]. The ribosomes were disrupted by adding an equal volume of 2% SDS solution and RNA was purified with RNA Clean & Concentrator-25 kit (“Zymo Research”, USA, Cat. No. R1017) following the manufacture’s protocol for the isolation of RNA fragments of less than 200 nt long. Nucleic acids were subjected to electrophoresis in denaturing (7 M urea) 15% polyacrylamide gel in TBE-buffer (0.89 M Tris, 0.89 M boric acid, 2 mM EDTA) and visualized by placing gels on a SkyLight ECX-F20 V1 transilluminator (“Vilber”, France) after staining with the SYBR Gold fluorescent dye (“Thermo Fisher Scientific”, Cat. No. S11494). A fragment of gel containing the band of putative RNA footprints was excised from the gel and passed through a hole in an Eppendorf tube by centrifugation to crash the gel. RNA was extracted from gel bits by incubating in nuclease-free water and precipitated in the presence of 0.2 µg/µl glycogen (Roche, Switzerland, Cat. No. 10901393001) and 0.3 M sodium acetate by adding 2 volumes of isopropanol. The RNA precipitate was pelleted by centrifugation, air-dried, and RNA was dissolved in nuclease-free water and stored at -80°C until further use. The preparation of the sequencing library, RNA sequencing, and read mapping were carried out by “Genoanalytics” LLC (Russia). The library was prepared with the NEBNext Small RNA Library Prep Set for Illumina (“New England Biolabs”, USA, Cat. No. E7330), following the manufacture’s protocol. The sequencing was conducted on an Illumina HiSeq 1500 system, using 75-nucleotides long single-end sequencing, with the output of 1 million reads. The reads were mapped to the human genome RESULTS AND DISCUSSION Fig. 1 shows results of electrophoretic analysis of the commercial DNA ladder as well as DNA and RNA oligonucleotides. As seen, under denaturing conditions, RNA oligonucleotides do migrate slower than DNA oligonucleotides with the matching length and nucleotide sequence (except for U and dT). The positions of 25 and 35 nt long RNA oligonucleotides on the gel coincide with those of 30 and 40 nt long DNA oligonucleotides of the commercial DNA ladder thus making them interchangeable for the RNA footprint identification purposes.
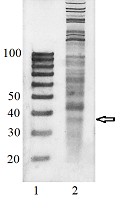
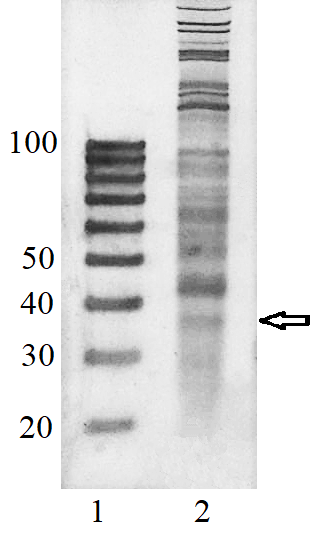
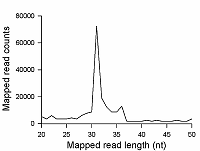
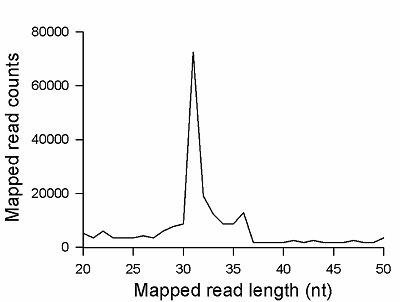
To verify that the 20/100 DNA ladder can be used to identify the RNA footprints on gels for further extraction, we subjected the RNA preparation enriched in the footprints to electrophoresis alongside with these DNA length standards. The results are shown in Fig. 2. The band which is positioned between 30 and 40 nt long DNA standards and expected to contain RNA footprints was excised, followed by RNA extraction and sequencing. The sequencing results are presented in Fig. 3 as a distribution of mapped read counts over their length. The distribution exhibited a maximum at the length of 31 nt that well agreed with the length of the RNA footprint of about 30 nt.
CONCLUSION The presented results demonstrate that the commercial 20/100 DNA oligonucleotide length standards can be successfully employed to identify the position of RNA footprints on denaturing polyacrylamide gels for the purpose of translatome profiling by RNA sequencing. The band composed of RNA footprints of about 30 nt long is positioned on gels between DNA size standards of 30 and 40 nt long. COMPLIANCE WITH ETHICAL STANDARDS This article does not contain any research involving humans or using animals as objects. FUNDING The study was performed within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021–2030) (No. 122030100170-5). CONFLICT OF INTEREST The authors declare no conflict of interest. REFERENCES
|