|
СОДЕРЖАНИЕ 1. АБИРАТЕРОН И ГАЛЕТЕРОН КАК “МУЛЬТИТАРГЕТНЫЕ” АГЕНТЫ 2. ПРОИЗВОДНЫЕ И АНАЛОГИ АБИРАТЕРОНА И ГАЛЕТЕРОНА 3. ПРОИЗВОДНЫЕ АНДРОСТАНА, СОДЕРЖАЩИЕ ГЕТЕРОЦИКЛ ПРИ C17 4. ПРОИЗВОДНЫЕ ПРЕГНАНА, СОДЕРЖАЩИЕ ГЕТЕРОЦИКЛ ПРИ C21 5. ПРОЧИЕ СТЕРОИДНЫЕ ПРОИЗВОДНЫЕ Таблица 1Биологическая активность соединений 296-309. |
Стероидные ингибиторы CYP17A1 – платформа для разработки новых противоопухолевых агентов
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича,
119121 Москва, ул. Погодинская, 10 стр. 8, *e-mail: alexander.misharin@ibmc.msk.ru
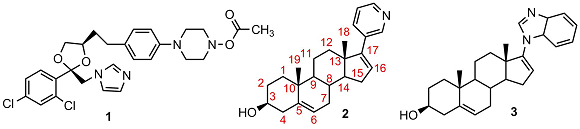
Ключевые слова: ингибиторы CYP17A1; абиратерон; галетерон; азотсодержащие стероидные производные; антипролиферативная активность; противоопухолевая активность DOI: 10.18097/BMCRM00020 ВВЕДЕНИЕ Поиск и разработка новых препаратов для борьбы с онкологическими заболеваниями является важнейшей задачей биомедицинской химии. В структуре смертности от онкологических заболеваний второе место занимает рак предстательной железы, характеризующийся резким повышением уровня андрогенов и гиперактивностью андрогенового рецептора. Еще в 40-е годы прошлого века была разработана стратегия лечения рака предстательной железы, в основе которой лежит снижение уровня андрогенов в простате [1,2]. Ключевым ферментом биосинтеза андрогенов является 17α-гидроксилаза-17/20-лиаза (СYP17A1) и, естественно, что ингибиторы CYP17A1 привлекали внимание исследователей в качестве потенциальных препаратов для лечения рака предстательной железы. Первым ингибитором CYP17A1, внедренным в клиническую практику, был противогрибковый препарат кетоконазол 1 [3]. Однако низкая специфичность и широкий спектр побочных эффектов этого препарата заставили специалистов начать поиск новых, более эффективных и специфичных ингибиторов CYP17A1. Mасштабные исследования, начатые в середине 90-х годов прошлого века, привели к созданию большой серии новых ингибиторов CYP17A1, которую принято условно подразделять на стероидные и нестероидные. Экспериментальные и клинические исследования стероидных ингибиторов CYP17A1: 17-(3-пиридил)-андроста-5,16-диен-3β-ола 2 (абиратерона) [4] и 17-(1H-бензимидазол-1-ил)-андроста-5,16-диен-3β-ола 3 (галетерона) [5] внесли существенный вклад в развитие фундаментальной онкологии. Абиратерон внедрен в клиническую практику в 2011 году в качестве препарата для лечения рака предстательной железы. Для стероидных ингибиторов CYP17A1 был разработан фармакофор [6], позволяющий предсказывать биологическую активность новых соединений. В 2012 году была определена пространственная структура комплексов CYP17A1 с абиратероном и галетероном [7]. Результаты исследований стероидных ингибиторов CYP17A1 и их использования в качестве противоопухолевых лекарственных препаратов неоднократно были обобщены в литературе ранее [8-16].
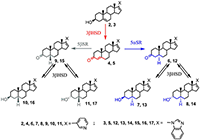
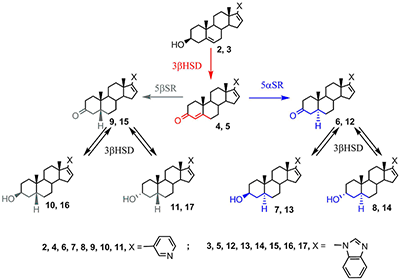
Поиск новых стероидных ингибиторов CYP17A1 продолжается в настоящее время, однако в этих исследованиях можно отметить новые тенденции. Если ранее основной целью работ был поиск наиболее эффективных и специфичных ингибиторов фермента, то в новых исследованиях основное внимание уделяется структурам, способным воздействовать сразу на несколько биологических мишеней, вовлеченных в патологический процесс. Для потенциальных лекарственных препаратов против рака предстательной железы – это в первую очередь андрогеновый рецептор, ферменты биосинтеза и метаболизма стероидов, а также компоненты регуляторных путей клеточного цикла, внутриклеточного сигналинга и апоптоза. В данном обзоре поставлена цель обобщения результатов исследований стероидных ингибиторов CYP17A1 и структурно родственных соединений, опубликованных в последнее десятилетие. В обзоре приведены структуры 354 новых соединений и данные об их биологической активности. В силу того, что в цитируемых работах определение биологической активности проводилось в разных моделях при помощи различных методов, количественные данные в обзоре не приводятся. При написании данного обзора нам хотелось подчеркнуть еще одну особенность разработки новых противоопухолевых препаратов на основе стероидных ингибиторов CYP17A1. Все экспериментальные исследования абиратерона и галетерона были проведены в академических и университетских лабораториях, а не в соответствующих подразделениях фармацевтических компаний. Исследования абиратерона, галетерона, их производных и аналогов внесли существенный вклад в развитие фундаментальной биомедицинской химии. Успех был достигнут не только благодаря высокопроизводительным технологиям, но и исследованиям биохимиков, химиков-синтетиков, онкологов, эндокринологов, молекулярных биологов. 1. АБИРАТЕРОН И ГАЛЕТЕРОН КАК “МУЛЬТИТАРГЕТНЫЕ” АГЕНТЫ Абиратерон и галетерон воздействуют на целый комплекс молекулярных мишеней, задействованных в жизнедеятельности опухолевых клеток, что позволяет использовать эти соединения в качестве агентов для подавления андроген-зависимых и андроген-независимых опухолей. Результаты многочисленных лабораторных и клинических исследований абиратерона показали, что СYP17A1 не является единственной биомишенью для этого соединения. Абиратерон ингибирует активность важнейших стероидогенных ферментов: ароматазы (CYP19), 11β-гидроксилазы (CYP11B1), альдостерон синтазы (CYP11B2), 3β-стероид-дегидрогеназы (3β-HSD), сульфотрансфераз SULT2A1, SULT2B1b и SULT1E1 [17-21]; он полностью подавляет экспрессию гена 21-гидроксилазы (CYP21A2) [22]. Прием абиратерона влияет на концентрацию глюкокортикоидов и минералокортикоидов [23], существенно влияет на профиль основных стероидов (метаболом) в организме [22]. Абиратерон в высоких концентрациях снижает уровень андрогенового рецептора и его активность [24,25], а также является умеренным антагонистом сплайс-вариантов андрогенового рецептора, характерных не только для карциномы простаты, но и для других андроген-зависимых опухолей, в том числе рака молочной железы [26] и яичников [27]. Галетерон подавляет активность CYP17A1 слабее, чем абиратерон, однако является мощным антагонистом андрогенового рецептора, его мутантных форм и альтернативных сплайс-вариантов, а также агентом, стимулирующим протеосомальную деградацию андрогенового рецептора (ARDA – andogen receptor degrading agent) [28]. Галетерон ускоряет деградацию полноразмерного рецептора (а также его мутантов, лишенных лиганд-связывающего домена), ингибируя активность USP12 (фермента системы деубиквитинилирования, который сам является также коактиватором андрогенового рецептора [29,30]). Абиратерон и галетерон способны влиять на механизмы регуляции клеточного цикла, внутриклеточного сигналинга и апоптоза. Обработка клеток LNCaP (андроген-зависимых) и PC-3 (андроген-независимых) абиратероном вызывала снижение уровня рецепторов TGFβ I и II и белков-посредников SMAD 3 и 4, - белков, вовлеченных в сигнальные пути цитокина TGFβ [31,32]. Cигнальный путь TGFβ играет важную роль в пролиферации опухолевых клеток, росте опухоли, и, особенно, возникновении метастаз. Абиратерон активирует белок p53 – транскрипционный фактор, выполняющий функцию супрессора образования злокачественных опухолей и проапоптотического фактора для «дефектных» клеток. Абиратерон подавляет экспрессию генов анти-апоптотических факторов - сурвивина, p21 и каспазы CASP3, одновременно повышая уровень аннексина V и степень его интернализации в мембрану клетки (что является достоверным маркером апоптоза). [33] В клетках карциномы простаты PC-3, LNCaP, CWR22Rv1 и карциномы поджелудочной железы MiaPaCa-2, S2-013, MiaPaCa-GR и MiaPaCa-GTR галетерон подавлял сигнальные пути Mnk1/2-eIF4E и NF-κB способствуя деградации киназы Mnk1. При этом наблюдалось снижение эспрессии генов Snail, Slug, N-кадгерина, виментина и MMP-2/-9, белковые продукты которых задействованы в миграции опухолевых клеток и, соответственно, в метастазировании [32-34]. Галетерон способен не только подавлять сигнальные пути, важные для роста, пролиферации и миграции опухолевых клеток, но и селективно активировать некоторые про-апоптотические факторы. Так показано, что галетерон способен повышать уровень про-апоптотических факторов Bax/Bcl2 и индуцировать апоптоз, опосредованный выходом цитохрома С в клетку [32-34]. Под действием стероидогенных ферментов и абиратерон, и галетерон подвергаются метаболическим превращениям. Основной метаболит абиратерона – 17-(3-пиридил)-андроста-4,16-диен-3-он 4 (D4A) был идентифицирован в плазме крови подопытных мышей, в плазме крови пациентов, принимавших ацетат абиратерона, и в клетках LNCaP, трансфицированных геном 3β-HSD [35]. Основной метаболит галетерона 17-(1H-бензимидазол-1-ил)-андроста-4,16-диен-3-он 5 (D4G) был идентифицирован в плазме крови подопытных мышей и клетках HEK293, трансфицированных генами стероидогенных ферментов [36]. Окисление абиратерона в D4A и галетерона в D4G происходит в присутствии 3βHSD, при этом абиратерон и галетерон, являясь конкурентными субстратами 3βHSD 1 и 2, эффективно подавляют превращение дигидроэпиандростерона в 3,17-андрост-4-ен-дион [37] (рис.1).
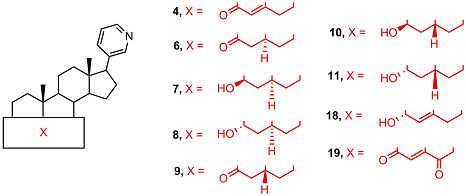
D4A 4 и D4G 5 не накапливаются в организме, а превращаются в насыщенные кетопроизводные 6, 9, 12, 15 под действием 5αSR и 5βSR, соответственно. Каждый из этих кетостероидов и в присутствии 3βHSD способен восстанавливаться до 3β- или 3α-гидроксистероида (7, 8, 10, 11 и 13, 14, 16, 17, соответственно). Таким образом, при приеме абиратерона или галетерона в каждом случае образуется по крайней мере 6 новых биологически активных соединений. Метаболиты абиратерона и галетерона показали значительный фармакологический потенциал, а также продемонстрировали, что незначительные изменения в структуре могут привести к новым соединениям, биологическая активность которых существенно отличается от таковой для абиратерона и галетерона. В настоящее время в лабораториях по всему миру проводятся работы по направленной химической модификации структур абиратерона и галетерона с целью получения новых аналогов и производных с улучшенной биологической активностью (подход, который в современной медицинской химии принято называть “оптимизацией структуры соединений-лидеров”). 2. ПРОИЗВОДНЫЕ И АНАЛОГИ АБИРАТЕРОНА И ГАЛЕТЕРОНА Исследование биологической активности и фармакологического потенциала метаболитов абиратерона и родственных соединений было проведено с использованием соединений 4, 6 – 11, 18 полученных встречным синтезом [35-39]. Соединения 4, 8, 9 связывались с CYP17A1 и ингибировали его активность аналогично абиратерону, а ингибиторная активность 3α-гидрокси-4-ен аналога 18 была ниже на 3 порядка [38]. Введение 6-кетогруппы существенно снижала ингибиторную активность - дикетостероид 19 слабо подавлял каталитическую активность CYP17A1 [40].
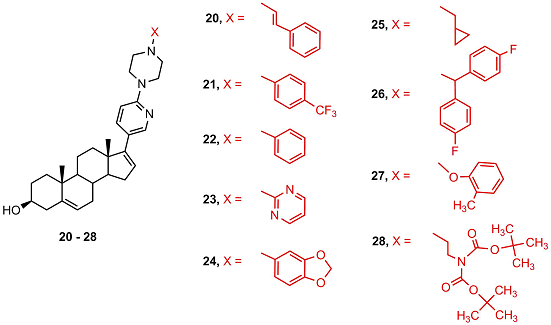
Метаболиты 4 и 9 обладали высоким сродством к андрогеновому рецептору дикого типа и мутантной форме Т877А, сродство абиратерона 2 и метаболита 8 было существенно ниже. Метаболит 9 активировал андрогеновый рецептор и стимулировал экспрессию рецептор-зависимых генов, подобно дигидротестостерону, в то время как метаболиты 8, 10, 11 не проявляли активности [35]. Метаболизм абиратерона 2 и D4А 4 с образованием 5α-H производных 6, 7, 8 вносит существенный вклад в противоопухолевую активность препарата; нежелательное превращение D4A в неактивные 5β-H производные 9, 10, 11 под действием стероид-5β-редуктазы печени подавлялось приемом дутастерида (препарата на основе 4-азапрегнена, избирательно ингибирующего этот фермент) [35,39]. В работе [41] были синтезированы и исследованы производные абиратерона 20 - 28, содержащие объемистые заместители в пиридиновом цикле.
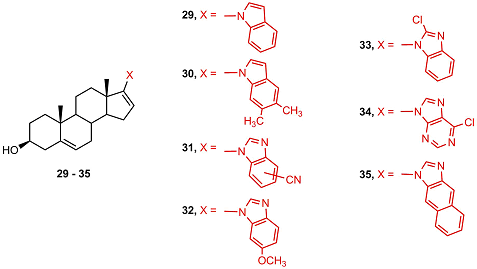
Соединение 23 избирательно подавляло активность CYP17A1, а соединение 26 – активность CYP19, однако ингибиторная активность была невысока. Молекулярный докинг показал, что связывание ингибитора 23 в активном центре CYP17A1 существенно отличалось от связывания абиратерона. Соединения 24, 26, 28 подавляли пролиферацию и стимулировали апоптоз только в гормон-независимых клетках карциномы простаты DU-145 и PC-3 [41]. С целью поиска новых антиандрогенов в работах [42,43] был проведен синтез новых аналогов галетерона, в которых последовательно варьировались структуры заместителей в положениях 17, 16, 3, а также изменялось количество и положение двойных связей в стероидном фрагменте.
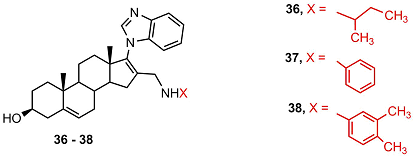
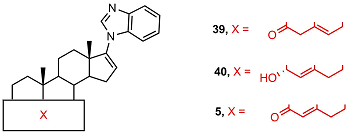
В ряду аналогов галетерона 29 - 35 не было выявлено соединений, обладающих сродством к андрогеновому рецептору, кроме того соединения 29, 30 и 35 стимулировали активность рецептора и не подавляли пролиферацию клеток LNCaP. Способность подавлять активность CYP17A1 у соединения 29 была ниже чем у галетерона в 200 раз. Введение в молекулу галетерона заместителей в положение 16 (соединения 36-38) приводила к полной потере противоопухолевой активности.
Антипролиферативная активность соединений Антипролиферативная активность соединений 39 и 40 была существенно ниже (в 5 раз), чем активность галетерона 3 и соответствующего 3-кето-4-ен-производного 5.
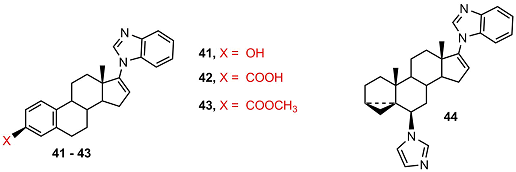
Аналоги галетерона, содержащие эстрановый фрагмент вместо андростанового (41-43), слабо подавляли стимулированную дигидротестостероном активность андрогенового рецептора, но кислота 42 вызывала быструю деградацию андрогенового рецептора.
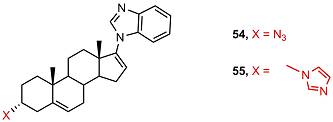
6-Замещенный имидазолид 44 эффективно подавлял стимулированную дигидротестостероном активность андрогенового рецептора, но активность этого соединения была значительно слабее, чем у 3-замещенного имидазолида 53.
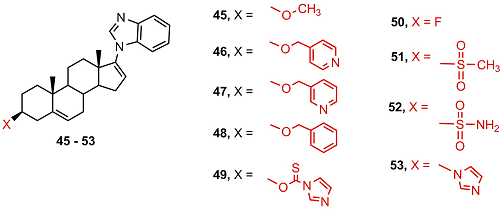
В ряду 3β-замещенных производных галетерона 45-53 соединения 49 и 53 полностью блокировали активность андрогенового рецептора, соединения 45 и 46 показали высокую активность, остальные соединения были неактивными. Соединение 46 сильно ускоряло деградацию мутантного андрогенового рецептора ARV7. Антиандрогенная и антипролиферативная активность 3α-имидазолида 55 была на порядок ниже, чем у соответствующего 3β-изомера 53.
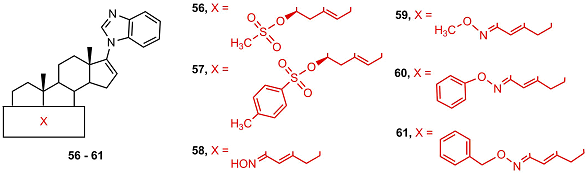
3β-Мезилат (56) и 3β-тозилат (57) галетерона слабо подавляли пролиферацию клеток LNCaP, а оксимы имели значительную антипролиферативную активность, которая убывала в ряду 58 > 59 > 60 (активность бензилоксима 61 не исследовалась из-за низкой растворимости).
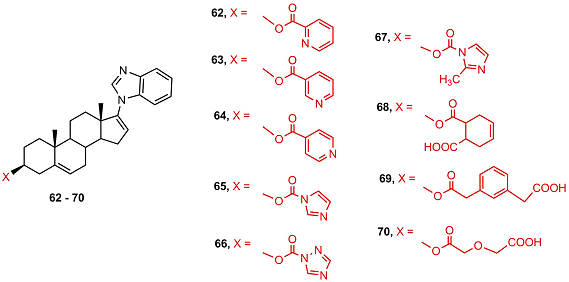
Ацилирование галетерона пиридинкарбоновыми кислотами приводило к соединениям 62-64, сильно подавляющим активность андрогенового рецептора и пролиферацию клеток LNCaP. Активность соединениния 62 соотвествовала активности галетерона. Самым сильным антиандрогеновым и антипролиферативным эффектом обладал имидазолилкарбамат 65, активность которого превосходила активность галетерона в 4 раза. Bведение заместителя в имидазольный цикл (67) или замена имидазольного цикла на триазольный (66) резко снижала активность. В результате авторы пришли к выводу, что модификация галетерона в положении 3β- остатком имидазолкарбоновой кислоты (65) или его превращение в (4-пиридил)метиловый эфир (47) – перспективный подход к получению новых противоопухолевых препаратов с оптимизированной структурой [42,43].
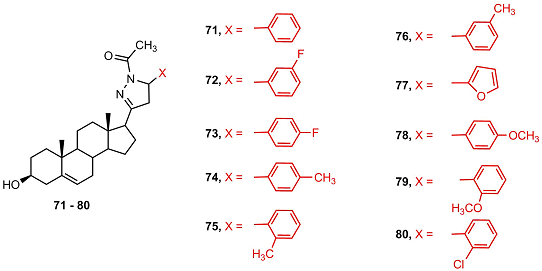
3. ПРОИЗВОДНЫЕ АНДРОСТАНА, СОДЕРЖАЩИЕ ГЕТЕРОЦИКЛ ПРИ C17 В рамках программ по поиску новых противораковых агентов в нескольких лабораториях проведён синтез новых стероидных производных, в структуре которых присутствовал арил-замещенный азотистый гетероцикл (пиразоловый, пиразолиновый, оксазоловый, оксазолиновый, оксазиновый, изоксазоловый) связанный с атомом С17 стероида. По сравнению с ингибиторами CYP17 A1, разработанными ранее [8-16], эти производные, как правило, слабее подавляли активность фермента, однако многие соединения этой серии проявляли высокую антипролиферативную активность в опухолевых клетках. В работе [44] был осуществлен синтез серии арил-замещенных стероидных пиразолинов 71-80 и проведен скрининг их биологической активности в культуре клеток карциномы кишечника (HT-29, HCT-15), легких (HOP-62, A-549), молочной железы (MCF-7) и глиобластомы (SF-295). Соединения 72, 73, 75 и 80 показали высокую токсичность для клеток HT-29 и HCT-15, а соединение 71 – в клетках MCF-7.
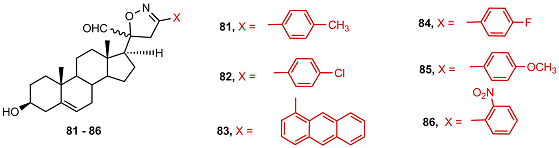
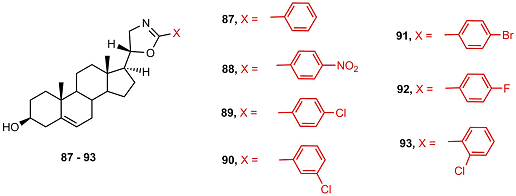
Стероидные производные, содержащие арил-замещенные изоксазолиновый (81-86) и оксазолиновый фрагменты (87-93), подавляли рост клеток карциномы простаты LNCaP, PC-3 и DU-145 [45].
Наибольшую антипролиферативную активность проявляли изоксазолины 81 и 85, содержащие электронодонорные заместители и оксазолин 87.
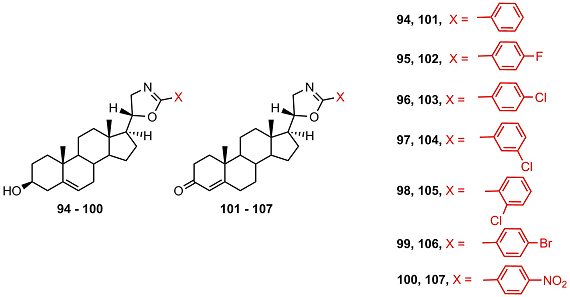
В работе [46] изучали эффекты арил-замещенных оксазолиновых производных 3β-гидроксиандрост-5-ена (94 - 100) и 3-кетоандрост-4-ена (101 – 107) на активность СYP17A1.
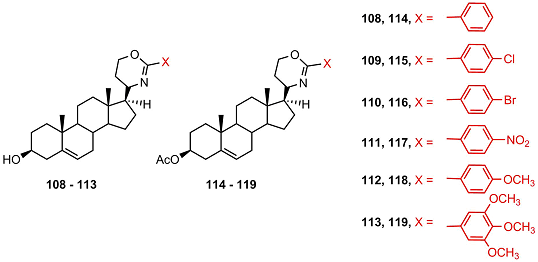
3β-Гидроксипроизводные не проявляли ингибирующей активности, а 3-кето-производные ингибировали активность этого фермента. Наиболее активными ингибиторами являлись кетостероиды 103 и 106, однако их активность была в 15 раз ниже активности кетоканазола. В отличие от оксазолиновых производных ни одно из оксазиновых производных (108 – 119) не обладало ингибиторными свойствами [47].
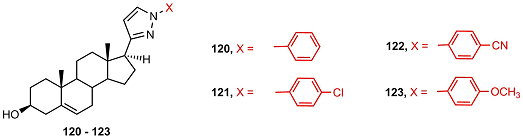
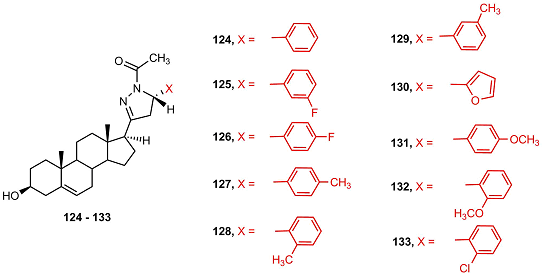
В работе [48] был проведен синтез арил-замещенных пиразолов 120-123 и пиразолинов 124-133. Синтезированные соединения исследовались в качестве ингибиторов 5αSR. Наиболее активными ингибиторами оказались соединения 121, 125 и 126 с галогенсодержащими заместителями, однако различия в ингибиторной активности были незначительными.
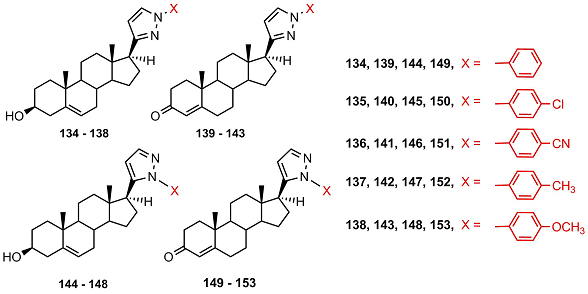
Производные 3β-гидроксиандрост-5-ена 134-138 и 144-148 не обладали ингибиторной активностью; среди производных 3-кетоандрост-4-ена активность фермента подавляли только 17β(1-цианофенил-3-пиразолил)андрост-4-ен-3-он 141 и 17β(1-п-метоксифенил-3-пиразолил)андрост-4-ен-3-он 143; активность наиболее сильного ингибитора (141) была слабее активности кетоканазола в 60 раз. Можно отметить, что соответствующие 5-пиразолил-содержащие 3-кетопроизводные 151 и 153 не являлись ингибиторами СYP17A1. Введение двойной связи в положение 16 стероида и замена 3β-гидрокси-5-ен- фрагмента на 3-кето-4-ен- незначительно увеличивали ингибиторную активность арилзамещенных пиразолинов [50].
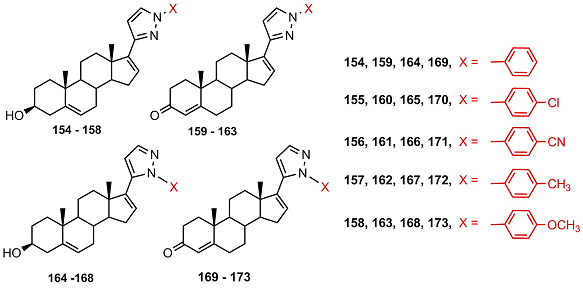
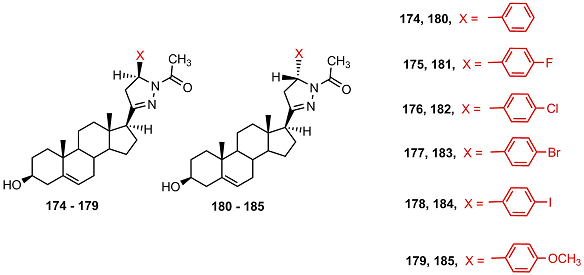
Среди Δ16- производных (154-173) соединения 169, 171 и 173 ингибировали активность СYP17A1, однако их активность была несопоставимо ниже, чем у незамещенных пиразолинов, исследованных ранее [11-13, 16]. В работе [51] были получены стероидные N-aцетилированные пиразолины, различающиеся конфигурацией атома С5’ в гетероцикле (174-185).
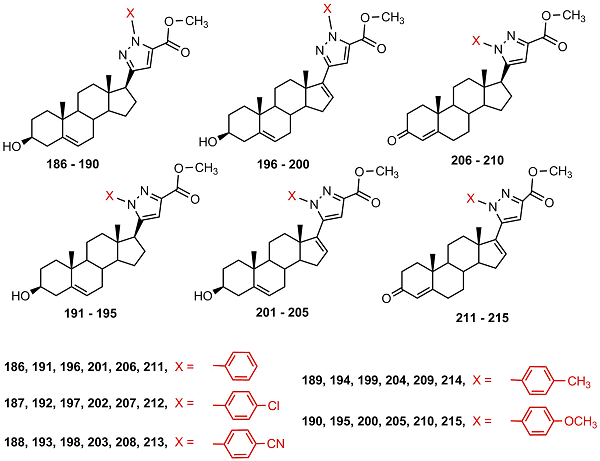
Ни одно из соединений не ингибировало активность СYP17A1; 5’S-изомеры сильнее подавляли рост клеток HeLa, MCF-7, A2780 и А431 по сравнению с соответствующими 5’R-эпимерами; 5’S-изомерные пиразолины 175 и 179 проявляли наибольший антипролиферативный эффект. В работе [52] были синтезированы стероидные пиразолы, содержащие ароматический заместитель при N1 или N2 и метоксикарбонильный заместитель при С’5 в гетероцикле (186-215). Синтезированные соединения были тестированы на способность подавлять активность CYP17A1, 3β-HSD и 17β-HSD3. Ни одно из соединений не подавляло активность CYP17A1, но 3β-гидроксистероиды 196, 200 и 3-кетостероид 206 эффективно снижали активность 3β-HSD. Среди вышеуказанных соединений были выявлены соединения с высокой антипролиферативной активностью в трех исследованных культурах клеток: HeLa, A431 и MCF-7 (199), a также с избирательной активностью в клетках HeLa (209). Антипролиферативная активность 3β-гидроксистероидов, содержащих насыщенный цикл D, была выше, чем у соответствующих Δ16-производных.
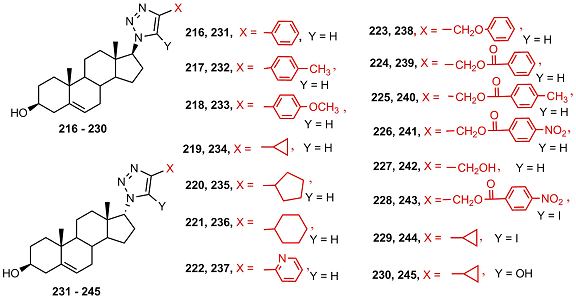
Изомерные стероидные триазолиды 216-245, различающиеся заместителями в гетероцикле и конфигурацией атома С17, изучались в качестве ингибиторов CYP17A1 [53].
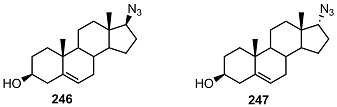
Среди исследованных соединений только 17α-изомерные триазолиды обладали ингибиторной активностью; наиболее сильными ингибиторами оказались соединения 240 и 245. Однако, ингибирующая активность 17α-триазолидов была на порядок ниже, чем у незамещенного 17β-азида 246. Изомерный 17α-азид 247 слабо ингибировал активность CYP17А1.
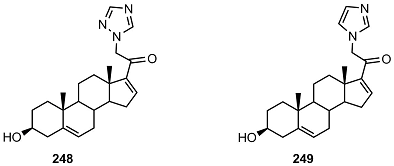
4. ПРОИЗВОДНЫЕ ПРЕГНАНА, СОДЕРЖАЩИЕ ГЕТЕРОЦИКЛ ПРИ C21 В работе [54] были синтезированы и исследованы 21-триазолил- и 21-имидазолил- замещенные производные 16-дегидропрегненолона 248 и 249.
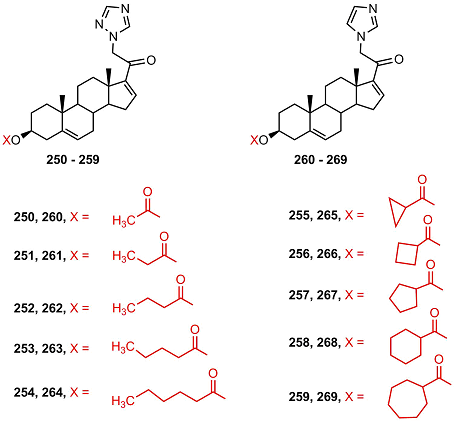
Оба соединения подавляли рост клеток РС-3, MCF-7, SK-LU-1 (карцинома легкого), но антипролиферативная активность имидазолида 249 во всех клетках была выше в 3 – 8 раз. Соединение 249 подавляло экспрессию генов циклинов D1 и E1 в клетках РС-3 и MCF-7, но не влияло на экспрессию Ki-67, EAG 1, BIM и сурвивина, обладало свойствами антагониста прогестеронового рецептора, но не имело антиандрогенной активности. Биологическая активность ацильных производных 21-триазолил- и 21-имидазолил- 16-дегидропрегненолона 250 – 269 изучалась в работах [55,56].
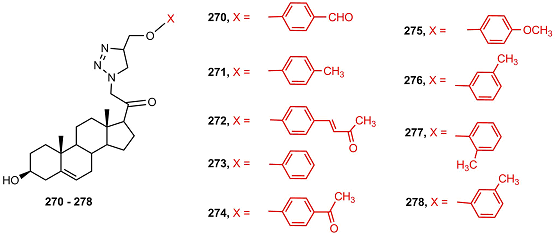
В ряду триазолидов эфиры 251 и 253 подавляли активность 5αSR 1 и 2; ацетат 250 обладал высокой цитотоксичностью в клетках SKLU-1; ацильные производные, содержащие остатки алициклических кислот 255-259, не обладали заметной биологической активностью. В ряду имидазолидов эфиры, содержащие остатки алициклических кислот, эффективно подавляли активность 5α-редуктазы 2, но не связывались с 5α-редуктазой 1 и андрогеновым рецептором; наибольшей активностью обладал эфир 268. Ацетат 260 обладал наиболее сильной антипролиферативной активностью в клетках РС-3, MCF-7 и SK-LU-1. Серия замещенных триазолил 20-кетопрегненов 270-278 была исследована на пролиферативную активность в культуре семи опухолевых клеток: PC-3, DU-145 (карцинома простаты), HCT-15, 502713 (карцинома кишечника), HEP-2 (карцинома печени), A-549 (карцинома легкого), SF-295 (глиобластома) [57].
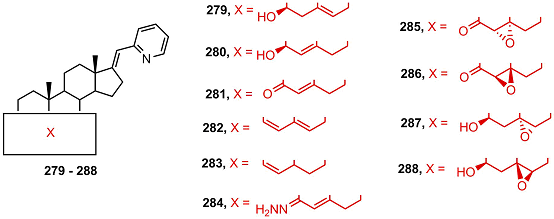
Соединения 270, 271, 272, 274, 276, 277 показали высокую антипролиферативную активность, причем соединение 274 было наиболее активно во всех тестируемых клетках. Исследования биологической активности и фармакологического потенциала (2-пиридил)-cодержащих производных [17(20)E]-21-норпрегнена 279-295 показали, что пиколиниденовые производные, содержащие 3β-гидрокси-5-ен- и 3-оксо-4-ен- фрагменты (279-281), эффективно ингибировали СYP17A1; активность убывала в ряду 281 > 279 > 280 [58]. Соединения 281, 283, 286 и 287 умеренно подавляли пролиферацию клеток карциномы простаты РС-3 и слабо – клеток карциномы молочной железы MCF-7 [59,60], однако соединения 279 и 282 сильно подавляли пролиферацию и вызывали апоптоз в клетках карциномы молочной железы MDA-MB-231 [61].
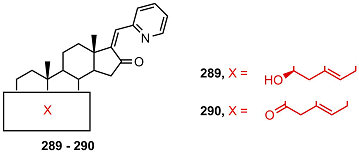
16-Кетоаналоги 289 и 290 подавляли активность СYP17A1 примерно с той же эффективностью, что и 16-незамещенные производные 279 и 281, и проявляли сильную токсичность в клетках миелоидного лейкоза линии K562 [62]; данные об активности в клетках карциномы простаты отсутствуют.
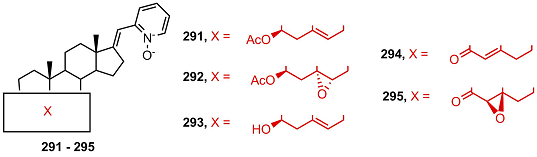
Высокую токсичность в клетках карциномы простаты РС-3 проявляли некоторые производные пиколинилиден-андростана, в которых атом азота был окислен до N-оксида, в частности ацетат 291. Интересно, что цитотоксичность соответствующего 3β-ацетилированного 5α,6α-эпоксида 292 была ниже в 100 раз, a соответствующего Δ5 стероида со свободной 3β-гидроксильной группой 293 – в 50 раз; антипролиферативная активность пиколинилиден-N-оксид-содержащих кетостероидов 294 и 295 в клетках PС-3 была умеренной и различалась незначительно [63].
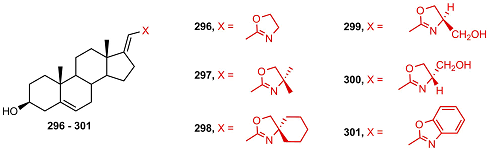
Оксазолин-содержащее производное [17(20)E]-21-норпрегнена 296 ингибировало каталитическую активность СYP17A1 сильнее, чем абиратерон; замена оксазолинового фрагмента на 4,4-диметилоксазолиновый (297) снижало ингибиторную активность на порядок, а замена на бензоксазоловый (301) – на два порядка [64-66]. Производные [17(20)E]-21-норпрегнена, содержащие полярные (299 и 300) или объемистые (298) заместители в положении 4 оксазолинового цикла, не связывались с ферментом и не проявляли ингибиторной активности [40,65].
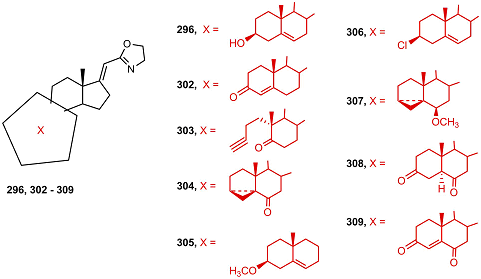
Среди аналогов оксазолина 296, различающихся структурой колец А и B, cильную ингибиторную активность проявляли 3-кето-4-ен- и секо-A- производные (302, 303). Удаление кислородсодержащей группы в положении 3, замена ее на хлор- или метоксигруппу, а также введение кетогруппы в положение 6 приводило к неактивным соединениям 305-309 [66].
Ингибиторная активность оксазолинов, а также их способность подавлять рост клеток карциномы простаты LNCaP и РС-3 снижались в ряду 296 > 303 > 302. (Таблица 1).
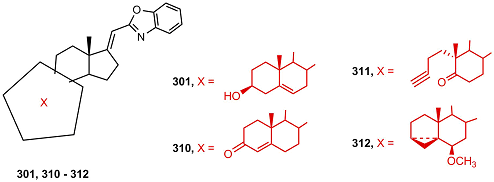
Бензоксазол 301, слабо ингибирующий активность СYP17A1, a также его аналоги 310 – 312, не подавляющие активность фермента, тем не менее, показывали антипролиферативную активность в клетках LNCaP и РС-3, сравнимую с таковой для абиратерона и оксазолинов 296, 302, 303 [66].
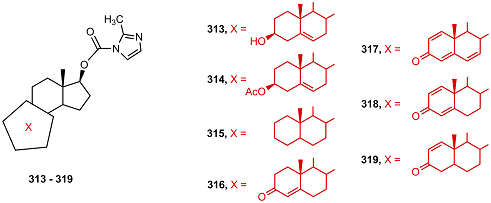
5. ПРОЧИЕ СТЕРОИДНЫЕ ПРОИЗВОДНЫЕ В работе [67] были синтезированы стероидные карбаматы 313 – 319. Полученные соединения были исследованы в качестве ингибиторов CYP17A1 и роста клеток РС-3, а также лигандов андрогенового рецептора. Cоединения 313 и 318 ингибировали активность CYP17А1, соединения 317 и 319 эффективно подавляли пролиферацию клеток PC-3, карбаматный фрагмент соединений 313-319 имел сродство к андрогеновому рецептору.
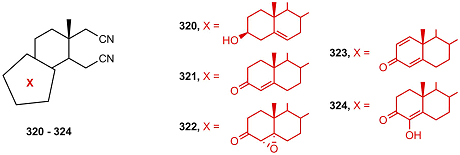
В работе [68] исследовалась серия стероидных 16,17-секо-динитрилов 320-324 в качестве ингибиторов пролиферации клеток MCF-7, MDA-MB-231 (карциномы молочной железы), PC-3 и HeLa (рак шейки матки).
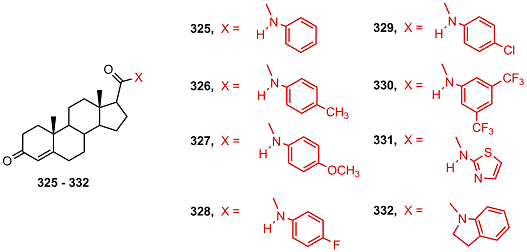
Соединение 323 подавляло пролиферацию клеток MCF-7 и MDA-MB-231 в субмикромолярных концентрациях. Молекулярный докинг позволил предсказать высокое сродство 16,17-секо-динитрилов к CYP17A1, ароматазе, андрогеновому рецептору, эстрогеновому рецептору α, AKR1C, 17β-HSD и/или 3β-HSD. В работе [69] исследован антипролиферативный и антиандрогеновый эффекты новых производных андрост-4-ен-3-она, содержащих арилкарбамоильную группу при С17 (325 – 332) в клетках LNCaP. Все соединения подавляли пролиферацию клеток, стимулированную тестостероном и дигидротестостероном; наибольшую активность проявляли соединения 330 и 331; эффект этих соединений был сильнее, чем у известных антиандрогенов финастерида, флутамида и кетоканазола. Амиды 330 и 331 также проявляли антипролиферативную активность в клетках РС-3 и не обладали острой токсичностью.
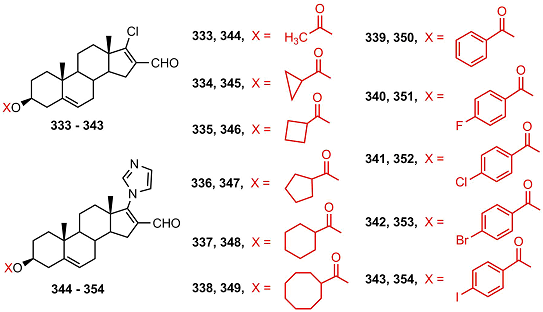
Исследование биологической активности новых производных дегидроэпиандростерона 333-354 [70] показало, что эти соединения являются довольно слабыми ингибиторами 5αSR 1 и 2 и не связываются с андрогеновым рецептором.
Тем не менее, стероиды 335, 351 и 352 показали антиандрогенную активность в экспериментах in vivo, сравнимую с финастеридом, а стероиды 342, 351 и 352 эффективно подавляли рост клеток PC-3. Результаты [70] указывают на высокий фармакологический потенциал производных дегидроэпиандростерона, ацилированных остатками ароматических кислот ЗАКЛЮЧЕНИЕ Необходимо отметить, что для разработки новых противоопухолевых агентов большой интерес представляет также направленная модификация стероидных ингибиторов других ферментов стероидогенеза, таких как 5α-SR [15,16,71,72] и 17β-HSD [73-78]. Природные соединения, в частности стероиды, дают уникальную возможность создания новых библиотек потенциальных лекарств [78-81], а накопленный специалистами огромный массив знаний о структуре и активности стероидов помогает быстро и эффективно проводить скрининг этих библиотек и поиск новых соединений-лидеров. БЛАГОДАРНОСТИ Работа выполнена при финансовой поддержке Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. ЛИТЕРАТУРА
|