|
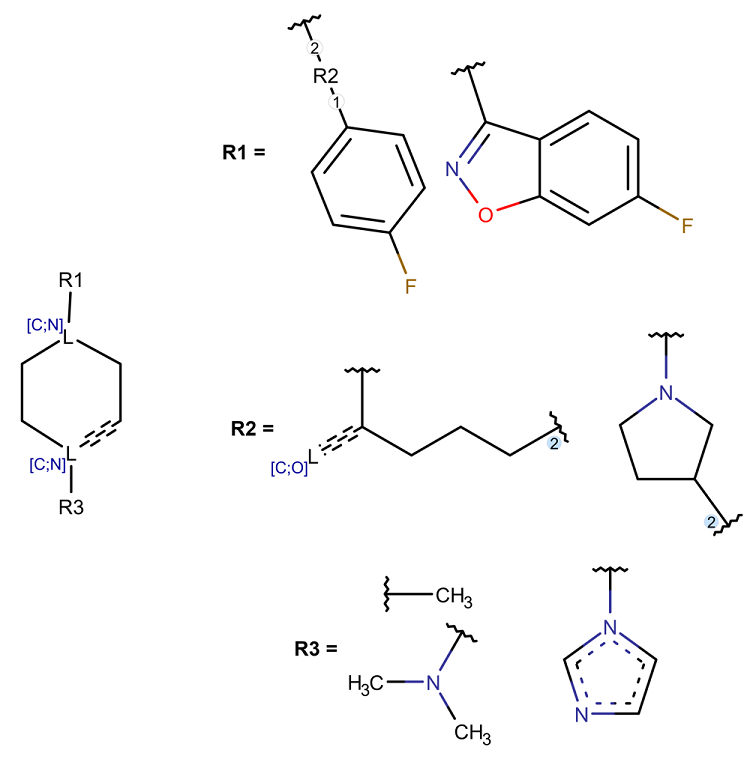
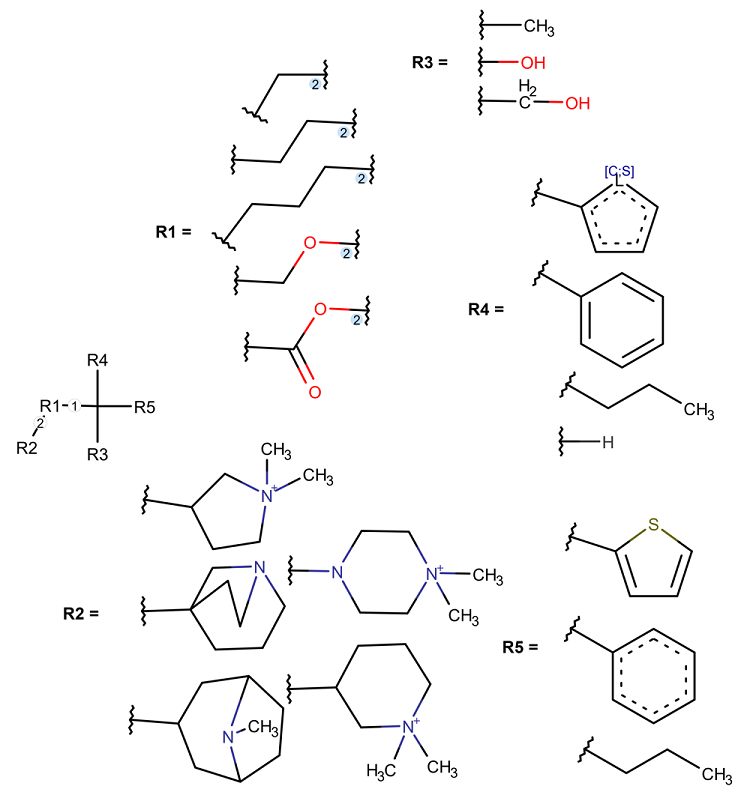
СОДЕРЖАНИЕ СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Рисунок 3Структурный фрагмент, характерный для ингибиторов ГМГ-КоА редуктазы. Рисунок 4Структурный фрагмент, характерный для ингибиторов EGFR. Рисунок 5Структурный фрагмент, характерный для антагонистов Д2Р. Рисунок 6Структурный фрагмент, характерный для М3Р. Таблица 1Характеристики основных этапов создания и валидации структурных фрагментов. |
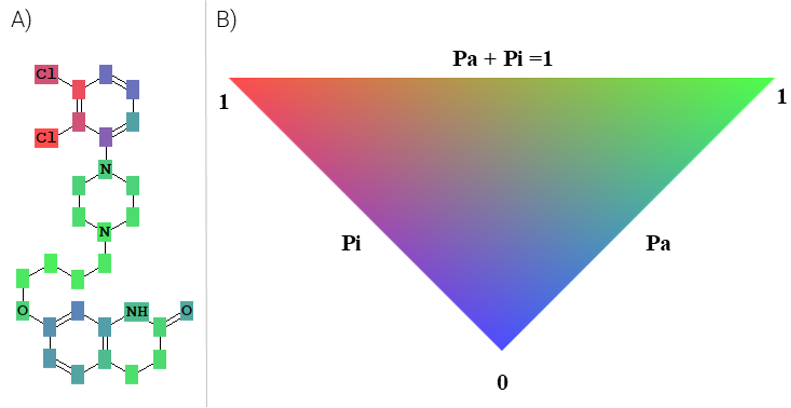
КОНСТРУИРОВАНИЕ ВЫСОКОСПЕЦИФИЧНЫХ СТРУКТУРНЫХ ФРАГМЕНТОВ ДЛЯ ФИЛЬТРАЦИИ СОЕДИНЕНИЙ С НЕЖЕЛАТЕЛЬНОЙ АКТИВНОСТЬЮ Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича, 112191, Москва, Погодинская ул., 10, стр. 8; *e-mail: polina.savosina@ibmc.msk.ru Ключевые слова: in silico исследования; оценка вклада атомов; структурные фрагменты; молекулярные мишени; нежелательные лекарственные реакции DOI: 10.18097/BMCRM00303 ВВЕДЕНИЕ Конструирование химических соединений с заданными свойствами является одним из основных направлений медицинской химии [1]. С одной стороны, для улучшения фармакодинамических и фармакокинетических характеристик лекарственной субстанции в ее структурной формуле проводят добавления или изменения структурных фрагментов. Однако внесение этих изменений требует обязательной последующей оценки полученных новых химических соединений для подтверждения их безопасности и эффективности перед дальнейшими исследованиями в клинических испытаниях. С другой стороны, доказательство безопасности лекарственной субстанции на ранних стадиях исследований может быть основано на результатах анализа ее вторичной фармакодинамики – взаимодействии с другими мишенями, отличными от терапевтической и называемыми также “off-target”. С этой целью проводят серии экспериментов с использованием широкого перечня молекулярных мишеней, относящихся к различным классам и играющих роль в разных физиологических процессах [2]. Выявление у исследуемого соединения активности по отношению к мишеням, ассоциированным с развитием нежелательных лекарственных реакций (НЛР), требует обязательной проверки полученных результатов in vivo; в случае их подтверждения может приниматься решение об остановке разработки этой субстанции и поиск более безопасной альтернативы [3]. В связи с этим целесообразно использовать предварительную оценку in silico для фильтрации наиболее опасных соединений и приоритизации дальнейших исследований на веществах с благоприятным профилем безопасности [4]. В настоящее время инструментом такой оценки может стать прогноз спектра видов биологической активности для химического соединения по его структурной формуле, который позволяет оценить вероятность наличия или отсутствия способности молекулы взаимодействовать с нежелательными мишенями. Однако его использование не всегда возможно, например по причине недостаточной точности прогностической модели, являющейся следствием высокого разнообразия положительных примеров в обучающей выборке. В подобных случаях методом выбора может стать оценка структурного сходства [5] или наличия/отсутствия структурного фрагмента, строго характерного для соединений, обладающих нежелательным видом биологической активности. Этот подход позволит еще на этапе конструирования химического соединения выполнять фильтрацию веществ, проявляющих нежелательные виды биологической активности, что значительно сократит как стоимость, так и время, необходимое для разработки нового лекарственного препарата. Целью нашей работы была разработка метода конструирования высоко специфичных структурных фрагментов, которые могут быть использованы для идентификации соединений, не обладающих нежелательными видами активности, на примере взаимодействия с молекулярными мишенями, ассоциированными с развитием НЛР. МЕТОДИКА Конструирование структурных фрагментов В качестве источника информации о взаимосвязях «структура-активность» мы использовали созданную нами ранее базу данных (БД) World Wide approved Drugs (WWAD), версия 2025.04 [6]. Она содержит информацию о структурных формулах, молекулярных мишенях, показаниях к применению и видах биологической активности для более чем 4500 низкомолекулярных активных фармацевтических субстанций (АФС), разрешенных к медицинскому применению. Информация о взаимодействии АФС с молекулярными мишенями собрана в WWAD из официальных документов, публикуемых 88 медицинскими ведомствами (общая характеристика лекарственного препарата, монография лекарства, инструкция по медицинскому применению, обзорные отчеты и пр.), и научных публикаций. Важно помнить, что при терапии одного заболевания нежелательным видом активности может являться такой эффект, который используется в качестве терапевтического для лечения другой нозологии. Это позволяет использовать базу данных WWAD для определения структурных фрагментов, специфичных как для терапевтических, так и для нежелательных эффектов. Для конструирования структурных фрагментов, характерных для конкретного вида нежелательной активности, мы использовали компьютерную программу PASS (Prediction of Activity Spectra for Substances), которая позволяет прогнозировать спектр биологической активности на основе структурной формулы исследуемого соединения [7]. В программе PASS биологическая активность химических соединений описывается качественно (“активно” или “неактивно”) и принимается, что соединение в обучающей выборке не обладает теми видами биологической активности, которые не указаны в списке его известных видов активности. Алгоритм построения зависимостей «структура-активность» для лекарственно-подобных соединений реализован в PASS на основе представления их структур специально разработанными дескрипторами MNA (многоуровневых окрестностей атомов) и модифицированного наивного Байесовского классификатора. Все соединения с определённым видом активности считаются положительными примерами, а все остальные – отрицательными. Это приближение не оказывает существенного влияния на результаты анализа зависимости «структура–активность» и выполняемого на этой основе прогноза благодаря тому, что алгоритм программы PASS обладает робастностью (статистической устойчивостью) по отношению к неполноте данных в обучающей выборке. Три положительных примера могут позволить получать статистически значимые оценки для такого вида активности. Результат прогноза PASS представляется в виде списка прогнозируемых видов биологической активности с соответствующими расчетными значениями вероятностей Pa («быть активным») и Pi («быть неактивным»). Атомно центрированные фрагментные дескрипторы MNA позволяют оценивать вклад в биологическую активность соединения каждого атома его молекулы. Для визуальной интерпретации рассчитанных вероятностей в программу добавлена цветовая индикация, в которой интенсивность красной составляющей цвета пропорциональна 0.3+0.7Pi, зелёной – 0.3+0.7Pa, синей – 1-0.7*(Pa+Pi). Таким образом, красный цвет соответствует вероятностям Pa=0 и Pi=1 (атом характерен для неактивных соединений), зеленый цвет – Pa=1 и Pi=0 (атом характерен для веществ, обладающих активностью), синий цвет – Pa=Pi=0 (вклад в проявление активности данного атома несуществен) (рис. 1). При низком уровне уверенности в прогнозе, например, Pa=Pi=0.3, цвет атома будет серым. Такая оценка позволяет выделять в структурной формуле соединения фрагменты, оказывающие положительное или отрицательное влияние на оценку его активности.
Разработанный нами метод конструирования структурных фрагментов можно представить в виде трёх этапов. На первом этапе с использованием специальной версии программы PASS Professional, которая позволяет создавать собственные SAR Base (база знаний о зависимостях «структура-активность»), на основе представленной в WWAD информации нами была построена собственная SAR Base для прогноза действия химических соединений на молекулярные мишени, ассоциированные с развитием НЛР. На втором этапе с использованием PASS выполнили прогнозирование НЛР только тех соединений, которые были использованы в качестве положительных примеров при создании SAR Base. На третьем этапе путем визуального сопоставления участков структур, окрашенных в зеленый цвет, создали структурный фрагмент (в виде т.н. структур Маркуша), общий для анализируемых соединений,. Для сохранения созданных структурных фрагментов в машиночитаемом формате Marvin Document (MRV) [8] и в формате изображений использовали программу MarvinSketch от компании ChemAxon. Валидация специфичности сконструированного фрагмента Подтверждение специфичности структурных фрагментов для требуемого вида биологической активности выполняли на основе БД PubChem, которая содержит информацию о более чем 122 млн химических соединений [9], в том числе о результатах биологического тестирования 4884139 веществ [10]. С использованием программного комплекса Instant JChem (IJC) от ChemAxon была создана локальная копия, содержащая структурные формулы и идентификаторы той части соединений PubChem, которая была связана хотя бы с одним результатом биологического тестирования, представленного в этой БД. С помощью имплементированного в IJC подструктурного поиска в локальной копии были выявлены соединения, содержащие в своей структуре хотя бы один из созданных фрагментов. Далее были проанализированы наименования молекулярных мишеней, в отношении которых найденные соединения были протестированы. Информация о веществах, для которых в PubChem имелись сведения об отсутствии активности в отношении требуемой нежелательной мишени, была проверена вручную. РЕЗУЛЬТАТЫ Для иллюстрации разработанного нами метода конструирования структурных фрагментов, которые могут быть использованы для фильтрации соединений с нежелательными свойствами, нами были выбраны следующие пять молекулярных мишеней, взаимодействие с которыми не только используется для терапии различных заболеваний, но и ассоциировано с развитием широкого спектра НЛР:
Важно упомянуть, что перечисленные НЛР могут возникать вследствие взаимодействия не только с выбранными биомакромолекулами. Множество других рецепторов и ферментов были определены в качестве потенциальных звеньев патогенеза этих НЛР с разной степенью достоверности [3]. Для каждой из этих мишеней в WWAD представлены взаимодействующие с ними АФС, что позволило с использованием PASS Professional на основе БД WWAD создать SAR Base, обеспечивающую прогноз описанных выше пяти механизмов действия с инвариантной точностью прогноза 0.9 и выше (табл. 1). Используя эту базу знаний, нами был выполнен прогноз для 5 наборов соединений, в каждом из которых присутствовали только АФС с требуемым видом активности. В результате мы получили изображения структурных формул веществ, на которых атомы были окрашены на основе их вклада в прогнозируемую активность с использованием цветовой схемы PASS (см. рис. 1Б). Пример таких изображений для ингибиторов АПФ представлен на рисунке 2А, для соединений из остальных групп – в Дополнительных материалах на рисунках S1-S4. Используя окрашенные в зеленый цвет участки структурных формул, мы построили структуры Маркуша, которые описывают важные для проявления активности участки структуры соединений.
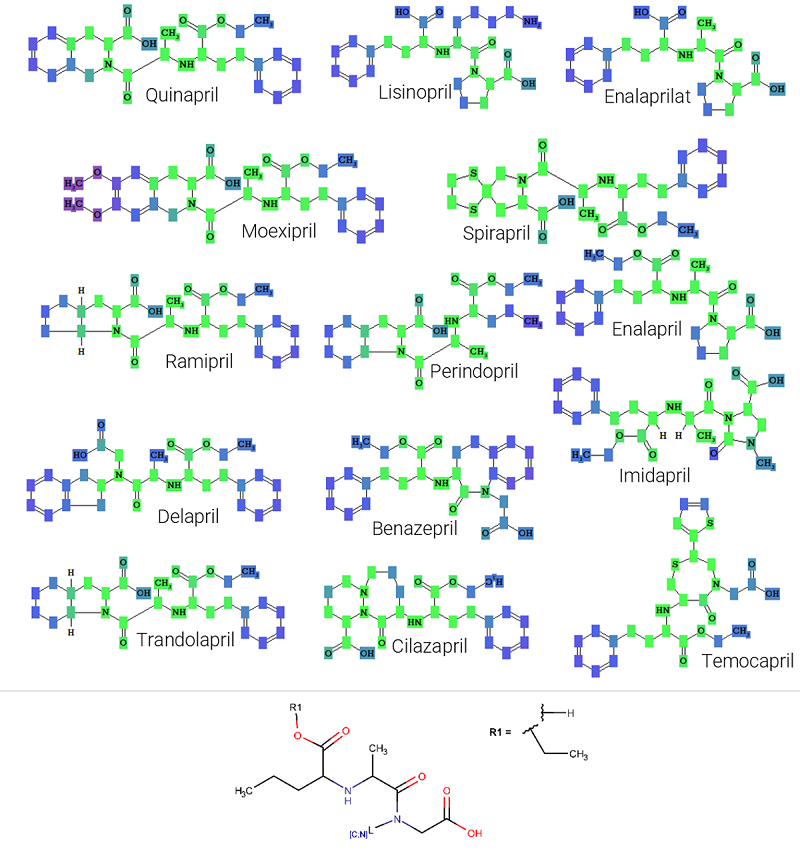
Ингибиторы АПФ I Первый структурный фрагмент был сконструирован на основе структурных формул ингибиторов АПФ (рис. 2). Используя его в качестве запроса с целью подструктурного поиска среди соединений PubChem, для которых существует информация о результатах биологических тестирований, мы идентифицировали 298 молекул. Для 187 из них способность ингибировать АПФ была подтверждена хотя бы в одном in vitro исследовании. Ни для одного вещества не было показано отсутствия анализируемой активности. Остальные 111 соединений не были исследованы на наличие АПФ ингибирующей активности.
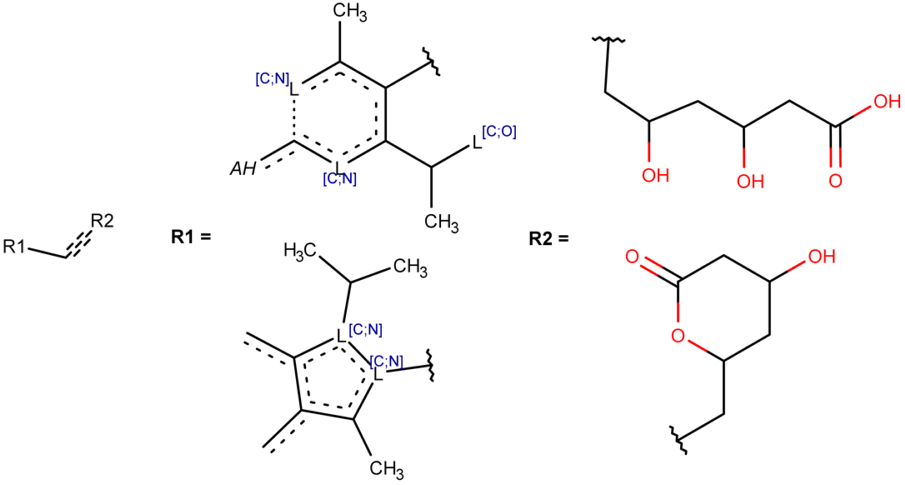
Ингибиторы ГМГ-КоА редуктазы Структурный фрагмент, характерный для ингибиторов ГМГ-КоА редуктазы (рис. 3), позволил найти в PubChem 458 соединений, для 257 из которых была подтверждена ингибиторная активность. Только для 4 структур наряду с положительными результатами тестирований присутствовали записи, в которых сообщалось об отсутствии активности в отношении ГМГ-КоА редуктазы. Среди них мы обнаружили разрешенное к медицинскому применению лекарство ловастатин, являющееся пролекарством, активный метаболит которого ингибирует анализируемую мишень, и два конъюгата, сконструированных из препарата аторвастатина, для его адресной доставки. Остальные 198 веществ по данным PubChem не были протестированы в отношении ГМГ-КоА редуктазы.
Ингибиторы EGFR Структурный фрагмент, специфичный для ингибиторов EGFR (рис. 4), присутствует в качестве подструктуры в 2988 соединениях PubChem. Наличие требуемой активности было подтверждено хотя бы в одном in vitro эксперименте для 1206 из них. Только для 20 найденных в PubChem веществ было упомянуто отсутствие ингибирования EGFR. При проверке первоисточников этой информации мы обнаружили, что в действительности эти 20 соединений могут блокировать действие требуемого тирозинкиназного рецетора, но их активность невысока (полумаксимальная ингибирующая концентрация превышает 10 мкМ). Для остальных 1762 веществ в PubChem не содержится записей о тестировании в отношении EGFR.
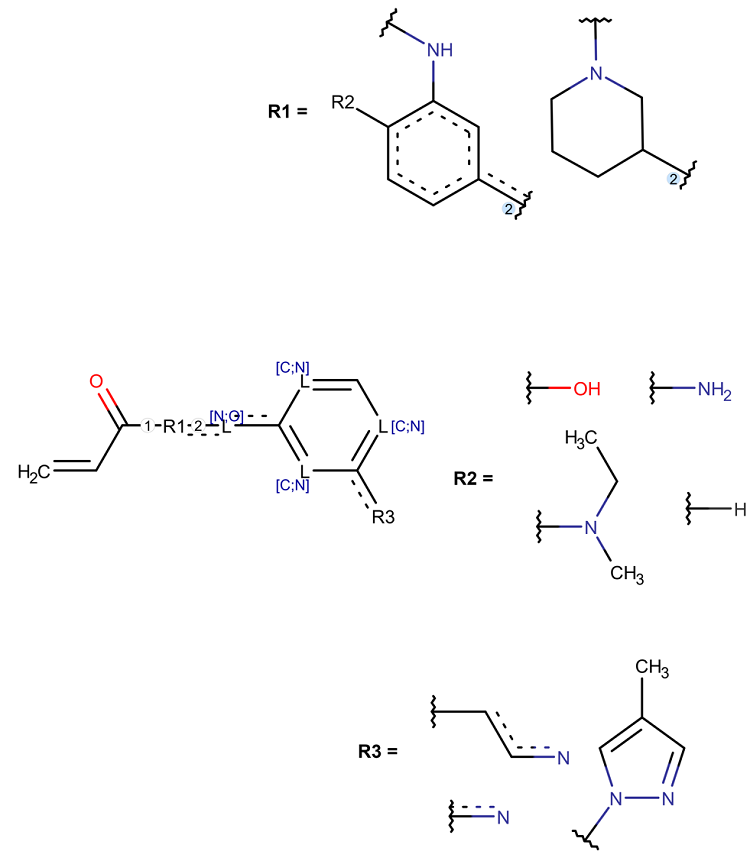
Антагонисты Д2Р Четвертый фрагмент, сконструированный для антагонистов Д2Р (рис. 5), позволяет найти 1471 соединение из PubChem. Записи с отметкой «Активен» в экспериментах по оценке способности связываться или блокировать Д2Р существуют для 603 найденных веществ. Неактивны как при анализе способности связываться с Д2Р, так и в функциональном тестировании в отношении этого рецептора 39 найденных структур. После проверки информации в первоисточниках мы обнаружили, что 6 молекул обладают невысокой активностью (константа диссоциации превышает 10 мкМ). Еще 33 вещества были неактивны по результатам высокопроизводительных скрининговых компаний, проведенных NCATS (PubChem AID 485344 и 624463), для которых известна высокая частота ложноотрицательных результатов. Например, в указанных отчетах о биологическом тестировании мы обнаружили отрицательные результаты для известных антагонистов Д2Р бенперидола, бромперидола, дроперидола, спиперона, галоперидола и др. Для остальных 829 из найденных соединений информация об экспериментальной оценке активности в отношении Д2Р в PubChem отсутствует.
Антагонисты М3Р С использованием пятого фрагмента, сконструированного для антагонистов М3Р (рис. 6), в анализируемой части PubChem было найдено 887 веществ. Активными хотя бы в одном экспериментальном тестировании по оценке связывания или наличия активности антагониста М3Р были признаны 274 молекулы. Ни для одного из найденных соединений не сообщалось исключительно об отрицательных результатах. Для остальных 613 веществ информация об экспериментальной оценке активности в отношении М3Р в PubChem отсутствует.
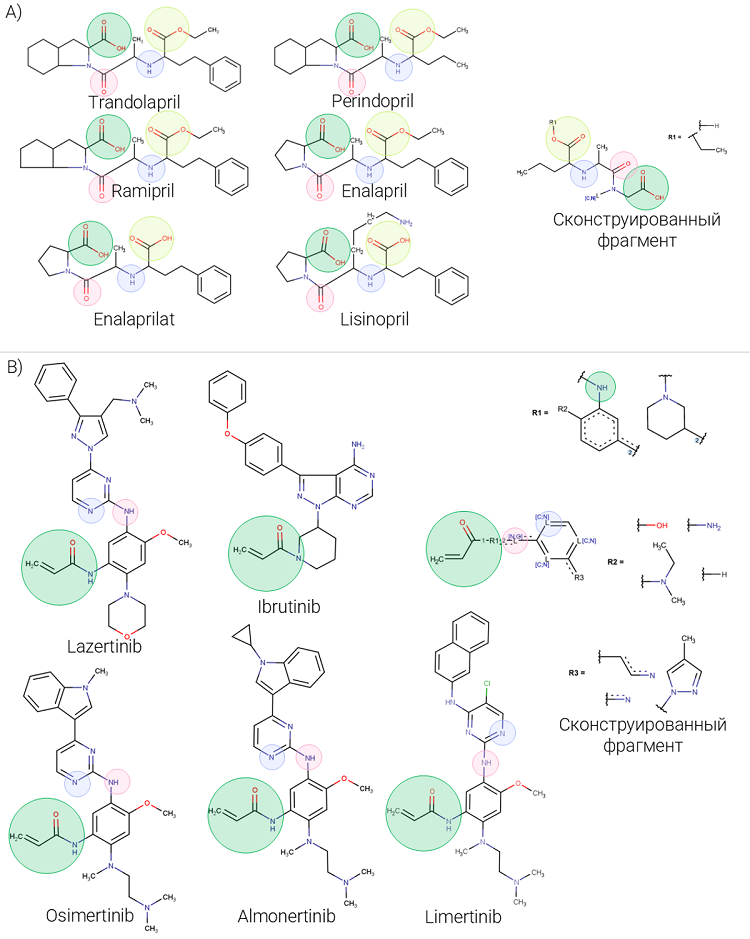
ОБСУЖДЕНИЕ С использованием разработанного нами метода на основе оценки вкладов отдельных атомов в проявление нежелательных видов активности нам удалось сконструировать характерные для них структурные фрагменты. Предварительное создание SAR Base позволило выявить не просто общие для всех анализируемых соединений участки структуры, а наиболее часто встречающиеся атомы с учетом их ближайшего окружения, которые с высокой вероятностью важны для проявления требуемого вида активности. В этом состоит преимущество предложенного нами подхода конструирования высоко специфичных структурных фрагментов по сравнению, например, с поиском максимально общей подструктуры [21] для соединений с одним видом активности. Молекулярные мишени, выбранные для демонстрации разработанного нами подхода, не только используются для достижения терапевтического эффекта, но и включены в панели фармацевтических компаний для оценки вторичной фармакодинамики и безопасности разрабатываемой терапии [3, 22]. Составленные нами структурные фрагменты являются более специфичными по отношению к конкретным молекулярным мишеням и НЛР, чем предложенные ранее другими авторами (например, так называемые PAINS фильтры [23]). Эти фрагменты позволяют не просто определить наличие опасной функциональной группы в структурной формуле разрабатываемого соединения, но и точно указать, взаимодействие с какой мишенью определяет потенциальный риск. Важно отметить, что сконструированные нами in silico высоко специфичные структурные фрагменты включают в себя участки структуры молекул, которые, по результатам исследования трехмерных комплексов «мишень-лиганд», отвечают за их формирование. Например, все функциональные группы, обеспечивающие связь ингибиторов АПФ с этим ферментом [24], представлены в определенном нами фрагменте (рис. 7А). Аналогичная зависимость была обнаружена при поиске информации о способе связывания некоторых ингибиторов c EGFR (рис. 7В) [25-27].
Оценка присутствия сконструированных фрагментов в структурах фармацевтических субстанций, одобренных к медицинскому применению, показала их уникальность для соединений с определенным видом активности, несмотря на то, что для их создания были использованы структурные данные и результаты прогноза лишь части имеющихся данных. Подструктурный поиск в более широком химическом пространстве PubChem, содержащем 4884139 экспериментально протестированных веществ, подтвердил высокую специфичность сконструированных фрагментов в отношении анализируемых молекулярных мишеней. Только 0.006-0.061% соединений содержали в своих структурных формулах построенные фрагменты. Однако менее половины веществ из найденных для каждого структурного фрагмента были экспериментально проверены на наличие требуемого вида активности. В то же время среди протестированных молекул либо все, либо подавляющее большинство взаимодействовали с мишенью, которая входила в список биомакромолекул, ассоциированных с анализируемой активностью. Важно подчеркнуть, что в разработанном нами подходе для поиска фрагментов были использованы данные исключительно о фармацевтических субстанциях, разрешенных к медицинскому применению. С одной стороны, это является недостатком данной работы, поскольку пространство одобренных АФС структурно менее разнообразно, чем все синтезированные на сегодняшний день вещества. В то же время, активность АФС была подтверждена на всех уровнях экспериментальных исследований (от биохимических тест-систем до организма человека), что исключает использование в предложенном подходе соединений, информация об активности которых является следствием ложноположительного результата тестирования. Ранее группой исследователей из США также был разработан подход для определения структурных фрагментов соединений, связанных с возникновением различных нежелательных реакций [4]. Этот подход был основан на выявлении взаимосвязей «структура-активность» с использованием методов глубокого обучения. В качестве обучающего набора данных выступала информация о НЛР одобренных АФС из БД SIDER, в которой с использованием методов автоматизированного анализа текстов собрана информация из официальных документов (сводных характеристик лекарственных препаратов, инструкций по медицинскому применению лекарственных препаратов) медицинских агентств США, Канады, Австралии, Великобритании и Европейского союза. Авторами были построены модели, позволяющие выполнять прогноз различных НЛР с средней точностью от 0.852 до 0.977, а также идентифицированы участки структурных формул соединений обучающей выборки, определенные во время обучения как вносящие наибольший вклад в прогнозируемые виды активности. Позднее, исследователями из Китая также было опубликован алгоритм прогноза некоторых видов токсичности для исследуемых веществ, который позволяет получать информацию о структурных участках, связанных с проявлением анализируемой активности [28]. Для этого авторами были созданы специальные обучающие наборы данных, содержащие информацию об АФС, одобренных к медицинскому применению в США и проявляющих один из трех видов неблагоприятных эффектов: тератогенность, удлинение интервала QT на электрокардиограмме или рабдомиолиз. Авторами были построены три модели с использованием сверточных нейронных сетей, выполняющие прогноз указанных видов токсичности. Последующий анализ позволил идентифицировать участки структурных формул соединений из обучающих выборок, которые в процессе обучения были отмечены как вносящие наибольший вклад в проявление прогнозируемого эффекта. Эти участки представляют собой отдельные атомы или функциональные группы размером до 3 неводородных атомов. Предложенный нами метод имеет ряд преимуществ по сравнению с найденными аналогичными работами. Во-первых, в качестве обучающего набора данных мы использовали БД WWAD, в которой представлено большее количество АФС, разработанных и используемых в намного большем числе стран. Это позволяет охватить более широкое фармакотерапевтическое химическое пространство при конструировании фрагментов. Во-вторых, в WWAD данные о взаимодействии АФС с молекулярными мишенями собраны посредством ручной курации, что обеспечивает высокую достоверность используемых для обучения сведений в отличие от источника информации группы исследователей из США и Китая, для создания которого были применены методы автоматизированного анализа текстов. Помимо этого, наивный Байесовский классификатор, который лежит в основе использованной в нашем методе программы PASS, обладает меньшей вычислительной сложностью и обеспечивает более высокую точность прогноза по сравнению с методами глубокого обучения, примененных в работах сравнения. Дополнительным преимуществом нашего подхода перед алгоритмом исследователей из Китая является получение более протяженных структурных фрагментов, ассоциированных с проявлением анализируемого вида биологической активности. ЗАКЛЮЧЕНИЕ Разработанный нами метод конструирования структурных фрагментов, высокоспецифичных для проявляющих определенный вид биологической активности веществ, может быть использован для фильтрации соединений с нежелательным типом активности, в том числе с целью разработки новых лекарственных средств. С помощью самого большого из находящихся в свободном доступе репозитория результатов биологических тестирований PubChem мы продемонстрировали, что построенные структурные фрагменты обнаруживаются в соединениях, для которых было экспериментально подтверждено взаимодействие с нежелательной мишенью. Приведенные в качестве примера молекулярные мишени, ассоциированные с различными НЛР, присутствуют в наборах для исследования вторичной фармакодинамики различных фармацевтических компаний. Это подтверждает актуальность и применимость предлагаемого нами подхода в решении практических задач, возникающих в ходе поиска новых лекарственных средств. Сравнение сформированных фрагментов с известными данными об участках структур молекул, ответственных за связывание с исследуемыми ферментами, указывает, что разработанный нами алгоритм оценки вклада отдельных атомов в проявление конкретного вида биологической активности может быть использован для определения функционально важных структурных участков соединений, которые взаимодействуют с молекулярными мишенями, трехмерная структура которых остается пока не охарактеризованной. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ В данной работе исследования с участием людей или использованием животных в качестве объектов не проводились. БЛАГОДАРНОСТИ Авторы выражают искреннюю признательность компании ChemAxon за предоставление лицензии на программный комплекс Instant JChem. Вычисления выполнены на оборудовании ЦКП «Протеом человека» НИИ биомедицинской химии имени В.Н. Ореховича. ФИНАНСИРОВАНИЕ Работа выполнена в рамках Программы фундаментальных научных исследований в Российской Федерации на долгосрочный период (2021-2030 годы) (№ 122030100170-5). КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов. ЛИТЕРАТУРА
|