|
Компьютерное прогнозирование спектров биологической активности химических соединений: возможности и ограничения 1Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121 Москва, ул. Погодинская, 10 стр. 8, *e-mail: vladimir.poroikov@ibmc.msk.ru 2Российский национальный исследовательский медицинский университет имени Н.И. Пирогова Минздрава России, 117997, Москва, ул. Островитянова, д. 1. Ключевые слова: анализ зависимостей «структура-активность», спектр биологической активности, компьютерное прогнозирование, PASS, точность, предсказательная способность, веб-ресурс Way2Drug DOI: 10.18097/BMCRM00004 ВВЕДЕНИЕ Наличие у химического соединения биологической активности дает возможность использовать его в качестве субстанции лекарственного препарата для терапии определенной патологии [1]. С другой стороны, биологическая активность может стать причиной проявления веществом побочных и токсических эффектов, что ограничивает возможности его практического применения [2]. В настоящее время около 80 млн различных химических соединений доступно для тестирования в виде уже синтезированных образцов [3]; в регистрационной системе Chemical Abstracts Service содержится информация о 138 млн органических и неорганических веществ, описанных в литературе с начала XIX столетия [4]; in silico сгенерированы сотни миллионов структурных формул органических молекул вместе с исходными реагентами и реакциями синтеза [5] и свыше 166 млрд структурных формул, полностью покрывающих химическое пространство, включающее до 17 атомов C, N, O, S и галогенов [6]. Число известных молекулярных мишеней лекарственных препаратов в организме человека составляет несколько тысяч [7, 8], а число фармакотерапевтических эффектов, возникающих при взаимодействии фармакологических веществ с этими мишенями – несколько сотен [9]. Экспериментальное тестирование взаимодействия многих миллионов химических соединений с тысячами молекулярных мишеней невозможно как с экономической, так и с практической точки зрения [10]. Таким образом, возникает необходимость предварительного отбора молекул, с наибольшей вероятностью взаимодействующих с целевыми молекулярными мишенями и проявляющих, благодаря такому взаимодействию, необходимое фармакотерапевтическое действие. С этой целью сегодня широко применяют компьютерные методы дизайна лекарств, основанные как на структуре макромолекулы-мишени, так и на структуре лигандов [11, 12]. Использование методов, основанных на структуре мишени, требует наличия информации о пространственной структуре макромолекулы-мишени, сопряжено с необходимостью проведения ресурсоемких вычислений и связано с рядом других ограничений [13]. В Интернете доступны некоторые веб-ресурсы, позволяющие предсказывать профили биологической активности на основе молекулярного докинга и анализа ассоциативных взаимосвязей по аффинности к различным молекулярным мишеням лекарственных соединений из обучающей выборки (например, [14]). Условием применения методов, основанных на структуре лигандов, является наличие «обучающих примеров» в виде массива информации о структуре молекул набора соединений и их взаимодействии с целевой мишенью или проявлении ими заданной биологической активности в некотором стандартизированном биологическом тесте. Часто это условие невыполнимо, особенно для новых фармакологических мишеней, представляющих особый интерес. В этих случаях применяют методы оценки биологической активности соединений на основе анализа структурного сходства их молекул, что не всегда гарантирует получение надежных оценок [15, 16]. Тем не менее, поиск «по сходству» является встроенной процедурой в некоторых базах данных коммерчески доступных образцов органических соединений (например, [3]), что помогает пользователю найти «хоть что-нибудь похожее» на структурную формулу, использованную в качестве запроса. Появление свободно-доступных через Интернет баз данных, содержащих информацию о структуре и биологической активности химических соединений [17-19], создало необходимые предпосылки для развития методов дизайна лекарств, основанных на структуре лигандов и позволяющих прогнозировать профили биологической активности для новых веществ [20-25]. Для прогноза биологической активности используется либо анализ структурного сходства (например, SwissTargetPrediction), либо методы машинного обучения (например, ChemProt). Программа PASS (Prediction of Activity Spectra for Substances) была создана намного раньше, чем упомянутые выше разработки, что было отмечено в недавней публикации Андреаса Бендера с сотр.: «One of the earliest and most widely used examples of data-mining target elucidation is the continuously curated and expanded Prediction of Activity Spectra for Substances (PASS) software, which was assimilated from the bioactivites of more than 270,000 compound-ligand pairs» [26]. В 1990 году была опубликована первая работа, в которой упоминалось о реализации нами прогнозирования спектров биологической активности [27]. В 1993 году были продемонстрированы преимущества программы PASS в сравнении с предсказаниями специалистов-экспертов для независимой выборки веществ [28]. Двумя годами позже было опубликовано детальное описание используемого в PASS подхода, включая некоторые примеры применения [29]. В 1996 году представлено первое описание PASS на английском языке [30]. В 1999 году был реализован первый в мире свободно-доступный веб-ресурс, позволявший пользователям осуществлять прогнозирование спектров биологической активности через Интернет [31, 32]. А в 2003 году результаты прогноза спектров биологической активности с использованием PASS для 250 тысяч соединений из базы данных Национального института рака США (Open NCI database) были представлены на веб сайте NCI/NIH [33]. Более детальную информацию об истории развития программы PASS можно найти в публикациях [34-37]. Ниже мы рассмотрим реализованный в настоящее время в PASS метод анализа зависимостей «структура-активность» и прогноза активности для новых веществ. МАТЕРИАЛЫ И МЕТОДЫ Биологическая активность органического соединения представляет собой результат его взаимодействия с биологическим объектом. Она зависит от характеристик соединения (структуры его молекулы), биологического объекта (вид, пол, возраст, и др.), способа воздействия (путь введения, доза) и особенностей условий эксперимента. В PASS биологическая активность описывается качественно (“активное” или “неактивное”); при количественных данных соединение признается «активным», если полуэффективная концентрация меньше 10 мкМ. Спектр биологической активности органического соединения – это множество различных видов биологической активности, которые отражают результат его взаимодействия с различными биологическими объектами. Он отражает “внутренние”, присущие данному соединению свойства, зависящие только от строения его молекулы. Вводя это обобщающее понятие, мы обеспечиваем возможность объединения больших массивов данных из различных источников, поскольку информация из конкретной публикации не охватывает всех аспектов биологического действия описываемого в нем органического соединения. При этом мы следуем принципу «презумпции невиновности»: в PASS принимается, что соединение не обладает теми видами биологической активности, которые не указаны в его спектре. Хотя нельзя исключить ситуации, когда информация о какой-либо активности органического соединения не была найдена в доступных источниках, либо оно обладает некоторой биологической активностью, но на эту активность соединение еще не испытывалось. Это приближение не оказывает существенного влияния на результаты анализа зависимости «структура–активность» и выполняемого на этой основе прогноза благодаря статистической устойчивости используемого в PASS метода расчета [38]. Прогнозируемый PASS спектр биологической активности органического соединения включает в себя фармакологические эффекты, молекулярные механизмы действия, специфическую токсичность и побочное действие, метаболизм, а также влияние на нежелательные мишени, молекулярный транспорт, генную экспрессию (рис. 1).
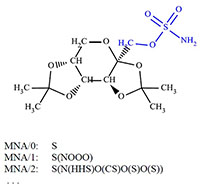
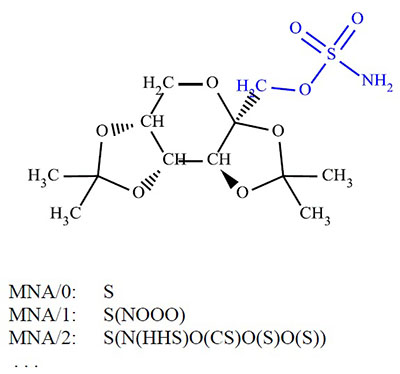
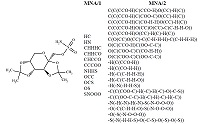
Необходимо подчеркнуть, что для прогноза с помощью PASS может быть использован любой способ объективной классификации органических соединений. Если соответствующие классы действительно определяются особенностями структуры молекул, то прогноз принадлежности к этим классам может быть вполне успешным. Например, интервал значений некоторой количественной величины можно рассматривать в PASS как «активность»: если значение величины принадлежит этому интервалу, то соединение «активно», и «неактивно» в иных случаях. Поэтому ясно, что применимость PASS гораздо шире прогноза только спектра биологической активности. Описание структуры органического соединения основано на его структурной формуле, так как это единственная доступная информация о соединении на ранних стадиях его исследования (соединение может только планироваться к синтезу). Для описания структуры химического соединения нами были предложены специальные дескрипторы, которые мы назвали дескрипторами MNA (Multilevel Neighborhoods of Atoms – многоуровневые атомные окрестности) [39]. Эти дескрипторы были разработаны с учетом нашего практического опыта по решению задач о поиске зависимостей «структура–свойство» для гетерогенных выборок органических соединений, обладающих широким набором видов биологической активности [34, 35]. Структурная формула, записываемая в соответствии с номенклатурными правилами в химии, отражает атомный состав и взаимное расположение атомов в молекуле. Дескрипторы MNA основаны на таком представлении структурной формулы, в котором, согласно валентностям и зарядам атомов, явно указаны все атомы водорода и не учитываются типы связей: природа не знает, что такое «стертые водороды», а кратность связей во многих случаях на самом деле должна быть дробной – например, в ароматическом кольце или в группе –NO2, – можно лишь утверждать, имеется ли между данными двумя атомами достаточно устойчивая химическая связь или нет. В таком виде структурная формула становится однозначной даже формально – она не зависит, например, от альтернативных способов изображения ароматических систем. На основе описанного представления структурной формулы дескрипторы MNA для каждого атома молекулы строятся рекурсивно следующим образом: дескриптор MNA 0-го уровня – метка A самого атома; дескриптор MNA любого следующего уровня – условное обозначение структурного фрагмента A(D1D2...Di...), где Di – дескриптор MNA предыдущего уровня для i-го непосредственного соседа данного атома с меткой A . Дескрипторы соседей D1D2...Di... записываются в каком-нибудь однозначном порядке, например, лексикографическом. Эта итерационная процедура может быть продолжена до любого уровня. Важно подчеркнуть, что метки атомов могут не только соответствовать общепринятым символам химических элементов, но и включать любую дополнительную информацию, например, о принадлежности атома к цепи или к какой-либо циклической системе, или что он является сайтом метаболизма. На рисунке 2 представлена структура молекулы противоэпилептического препарата Топирамат и пример дескрипторов MNA для атома серы. Структура молекулы в PASS представлена как бесповторное множество дескрипторов MNA 1-го и 2-го уровней. В дескрипторах 2-го уровня используется индикатор «–» для обозначения атомов, которые не входят ни в какие циклы. Пример множества дескрипторов MNA, описывающих в PASS соединение Топирамат, приведен на рисунке 3.
Оценить влияние стереоизомерии на проявление биологической активности PASS не позволяет, поскольку для многих видов активности в настоящее время невозможно создать репрезентативную обучающую выборку, учитывающую особенности пространственной структуры включенных в нее веществ. Оценка влияния на активность особенностей пространственной структуры изучаемых веществ может быть получена с применением методов молекулярного моделирования [40]. Важной особенностью дескрипторов MNA является их открытость – дескрипторы порождаются на основе самой структурной формулы, а не на основе какого-либо заранее составленного списка структурных фрагментов. Другая их особенность заключается в сохранении целостности фрагментов структуры в том смысле, что для каждого дескриптора MNA можно, при некотором навыке, изобразить соответствующий ему фрагмент. В программе PASS органические соединения считаются эквивалентными, если их молекулярные структуры описываются одинаковым набором дескрипторов. Так как дескрипторы MNA не отражают стереохимические особенности молекулы, структуры, которые отличаются только стереохимически, формально считаются эквивалентными. В PASS используются файлы, содержащие данные о химической структуре в форматах MOL или SDF [41]. Многие молекулярные редакторы и системы управления базами данных позволяют экспортировать данные в этих форматах. Дескрипторы MNA (как для прогнозирования спектра активности соединения, так и для добавления соединения в базу знаний с информацией о связи «структура–активность» SAR Base) генерируются только в случае, если структура соединения удовлетворяет следующим критериям:
Если структура не удовлетворяет этим критериям или имеются какие-либо другие ошибки во входных данных, то генерируется соответствующее сообщение об ошибке. Эти требования де факто являются стандартом, который в настоящее время общепринят и широко используется при подготовке обучающих выборок для построения зависимостей «структура-свойство» [42-44]. Обучающая выборка PASS состоит из тщательно отобранных записей о структуре и биологической активности органических соединений. Файл SAR Base (расширение «.SAR») создается в ходе обучения на основе обучающей выборки. SAR Base включает в себя словарь названий видов биологической активности (8054 терминов в версии 2017) и словарь дескрипторов MNA (106816 в версии 2017), данные, представленные в виде набора дескрипторов MNA соединений из обучающей выборки со спектрами их биологической активности (1025468 записей в версии 2017) и извлеченные в результате обучения знания о зависимостях «структура–активность» (7604 «правила» в версии 2017).К сожалению, пользуясь только публично доступными источниками, невозможно составить достаточно большую коллекцию биологически активных соединений, для которых были бы известны результаты тестирования на все виды биологической активности. По этой причине некоторые виды биологической активности в SAR Base PASS (версия 2017) представлены более чем 100000 органических соединений (136451 – противоопухолевые), а другие – только несколькими (405 видов активности по 3 соединения, 384 – по 4, 389 – по 5, и т.д.). В разных источниках информации биологические активности органических соединений описаны неодинаковыми терминами. Поэтому термины, описывающие спектры активности соединений в обучающей выборке, приводятся к единому «стандартизованному» виду. Алгоритм прогноза PASS удобнее всего описывать на основе классического байесовского подхода, который можно сформулировать следующим образом [45-47]. Для химического соединения C по структуре его молекул, записанной в виде множества {D1,...,Dm} из m дескрипторов MNA, оценим вероятность P(A|C) того, что соединение C принадлежит к классу A.Согласно формуле Байеса:
Если допустить, что дескрипторы D1,...,Dm независимы в совокупности, то можно, согласно «наивному Байесовскому подходу», записать P(C|A) как произведение условных вероятностей для отдельных дескрипторов:
Это выражение приближенное, поскольку дескрипторы MNA заведомо являются зависимыми в силу способа их построения. Но из-за отсутствия приемлемых альтернатив нам остается лишь не забывать о приближенности получаемых формул. В результате простых алгебраических преобразований получаем следующее выражение для логарифма отношения правдоподобия условной вероятности P(A|C) для класса A и P(B|C) для класса B в виде:
Смысл полученного выражения вполне прозрачен: логарифм апостериорного отношения правдоподобия есть сумма логарифма априорного отношения правдоподобия и суммы вкладов отдельных дескрипторов. При этом, если принадлежность к классам A и B не зависит от данного дескриптора, то P(A|Di)=P(A), P(B|Di)=P(B) и этот дескриптор не влияет на результат - его вклад в сумму нулевой. Это и есть результат классического байесовского подхода в приближении «наивный Байес» (Naïve Bayes) [45-47]. Формулу (3) можно записать в более традиционном виде. Для этого перенумеруем дескрипторы MNA по словарю и введем «координаты» соединений xi(C) так, что xi(C)=1, если дескриптор Di входит в описание соединения C, и xi(C)=0 во всех остальных случаях. Тогда формулу (3) для логарифма отношения правдоподобия принадлежности соединения C к классам A и B можно записать в следующем виде:
Формула (4) имеет вид обычной линейной регрессии, однако, коэффициенты регрессии ai в ней вычисляются не в результате минимизации какого-либо критерия согласия, а непосредственно на основе описанного байесовского подхода. В настоящее время в PASS класс B - это все соединения, не принадлежащие к классу A, P(B)=1-P(A),P(B|Di)=1-P(A|Di), и получаем из (3):
Алгоритм PASS использует частотные оценки вероятностей P(Ak) и P(Ak|Di) для класса Ak соединений, содержащих активность Ak в спектре активности:
где целые числа N ... Nik это: N - общее количество соединений в SAR Base; Ni - количество соединений, содержащих дескриптор Di в описании структуры молекул; Nk -- количество соединений, содержащих активность Ak в спектре активности; Nik -- количество соединений, содержащих и дескриптор Di в описании структуры молекул, и активность Ak в спектре активности. Помимо уже отмеченной приближенности подхода, формулы (3), (4) и (5) при использовании частотных оценок вероятностей (6) имеют существенный, хорошо известный недостаток -- оценки (6b) могут принимать нулевое значение. Один из наиболее популярных методов преодоления этого недостатка - «коррекция по Лапласу» [46, 47], согласно которому оценки (6) заменяются на следующие
Точность прогноза также повысилась после замены суммы вкладов дескрипторов их средним значением, что, видимо, компенсирует допущение о независимости дескрипторов. Для рассматриваемой задачи классификации логарифм априорного отношения правдоподобия (4b) не несет информации о конкретном прогнозируемом органическом соединении и может быть опущен. Описанный выше подход поясняет, почему алгоритм прогноза PASS основан на следующей специальной B статистике: по структуре молекул химического соединения, записанной в виде множества {D1,..., Dm} из m дескрипторов MNA, для каждой активности Ak подсчитываются величины Bk:
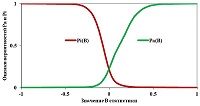
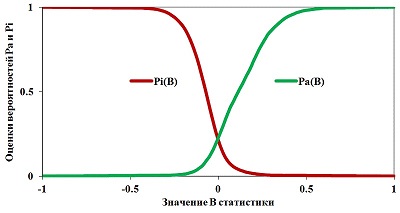
При этом для активности Ak, если для всех дескрипторов P(Ak|Di)=1, то Bk=1; если для всех дескрипторов P(Ak|Di)=0, то Bk=-1; если связи между дескрипторами и активностью Ak нет и P(Ak|Di) ≈ P(Ak), то Bk ≈ 0. Результат прогноза биологической активности представляется в PASS в виде вероятностей Pa «быть активным» («to be active») и Pi «быть неактивным» («to be inactive»). Зависимости, необходимые для получения вероятностей Pa и и Pi по значениям Bстатистики, и оценки точности прогноза PASS являются конечным результатом процедуры обучения, которая состоит в следующем. По данным SAR Base, сформированной на основе обучающей выборки, для каждой активности Ak для каждого из Nk активных и для каждого из N-Nk неактивных соединений вычисляются значения B статистики. Вычисления проводятся в режиме скользящего контроля с исключением по одному, т.е. после «исключения» этого соединения из SAR Base, для чего достаточно не включать его в суммы. По полученным выборкам B статистики строятся гладкие полиномиальные оценки функций Pa и Pi как описано в [38]. На рисунке 4 представлены Pa(B) и Pi(B) для активности «Антигипертензивное».
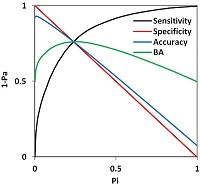
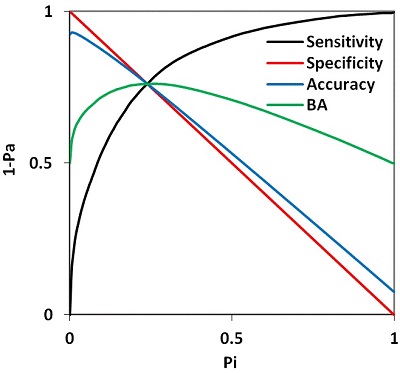
Из примера на рисунке 4 видно, что значения Pi монотонно убывают при возрастании значений Pa и сумма Pa и Pi меньше или равна 1. Вероятности Pa и Pi являются также, по построению, оценками вероятности ошибок прогноза 1-го и 2-го рода, соответственно, а 1-Pa и 1-Pi -- оценками чувствительности и специфичности. Вероятности Pa и Pi можно рассматривать и как меры принадлежности прогнозируемого соединения к нечетким множествам «активных» и «неактивных» органических соединений. Все эти интерпретации вероятностей Pa и Pi эквивалентны и полезны для анализа результатов прогноза. На их основе можно сконструировать самые разные критерии анализа результатов прогноза, соответствующие решению конкретных практических задач. Точность прогноза каждой активности оценивается в PASS как вероятность того, что для произвольной пары новых активного и неактивного соединений значение Pa для активного соединения будет выше, чем значение Pa для неактивного соединения, и называется инвариантной точностью прогноза (IAP) [35, 38]. Она эквивалентна критерию площади под кривой оперативной характеристики (AUC ROC). На рисунке 5 на примере активности «противоопухолевое» показаны зависимости между Pa и Pi, чувствительностью, специфичностью, точностью и сбалансированной точностью.
Площадь под кривой зависимости 1-Pa от Pi (кривой чувствительности), показанной на рисунке 5 черным цветом, и есть AUC ROC, совпадающая с IAP [35]. Точка пересечения всех кривых соответствует равенству Pa и Pi, и, соответственно, равенству вероятностей ошибок первого и второго рода, равенству чувствительности и специфичности, и, примерно, максимуму сбалансированной точности. Значение Pa = Pi в этой точке равно минимаксной оценке точности прогноза при полном отсутствии априорной информации как о платежной матрице, так и вероятности встречаемости активности в какой-либо выборке. Оценка влияния неполноты данных на качество прогноза. Поскольку обучающая выборка не может содержать полной информации о биологической активности включенных в нее соединений (ни одно из химических соединений не исследовано на все возможные виды биологической активности), мы провели специальное исследование [38] с целью оценки влияния неполноты информации в обучающей выборке на качество прогноза. Использовали выборку, содержащую около 19000 веществ из базы данных MDDR (так называемых “Principal Compounds”, для которых в MDDR были приведены экспериментальные данные о биологической активности). 120 различных видов активности было представлено в этой выборке не менее чем 100 соединениями. В ходе компьютерных экспериментов всю выборку 50 раз случайным образом делили на две равные подвыборки, одна из которых использовалась в качестве обучающей, а другая – в качестве тестовой, и наоборот (всего, таким образом, было выполнено по 100 экспериментов). Чтобы смоделировать неполноту информации, из обучающих выборок случайным образом исключали 20, 40, 60, 80% информации о структуре или биологической активности. В ходе обучения рассчитывали средние значения ошибки прогноза. Было показано, что исключение до 60% информации позволяет получать разумные оценки биологической активности для веществ тестовых выборок, то есть алгоритм программы PASS обладает робастностью (статистической устойчивостью) по отношению к неполноте данных в обучающей выборке. В данной работе также продемонстрировано, что оценка точности по скользящему контролю с исключением по одному даже более жесткая, чем по перекрестному контролю. Нами было проведено несколько компьютерных экспериментов по сравнению предсказательной способности PASS с другими, свободно-доступными через Интернет, веб ресурсами. В 2008 году отличных от PASS веб ресурсов в Интернете, прогнозирующих спектры биологической активности веществ, не было обнаружено; поэтому для сравнения качества прогноза мы сопоставили между собой результаты оценки некоторых других характеристик с применением различных методов [48]. Наилучшее согласие между результатами прогноза было получено для величины коэффициента распределения «н-октанол–вода» logP (для семи методов коэффициенты корреляции R варьировали от 0.65 до 0.90); менее согласованные между собой результаты были получены для прогноза растворимости в воде (R = 0.01 – 0.73 для четырех методов) и параметра «drug-likeness» (R = 0.19 – 0.73 для трех методов). Качество прогноза PASS было оценено на основе анализа независимых от авторов программы публикаций: было найдено 15 работ, в которых результаты прогноза были подтверждены в эксперименте для веществ, принадлежащих к различным химическим классам, и разнообразных видов биологической активности [48]. В 2016 году для веществ из тестовой выборки лекарственных препаратов, разрешенных к медицинскому применению в 2014 г., были проведены оценки качества прогноза с использованием четырех веб ресурсов [49]. Оказалось, что значения чувствительности S для четырех рассмотренных методов убывают в следующем порядке: PASS > SuperPred > DRAR-CPI > SwissTargetPrediction (S = 0.95; 0.53; 0.41; 0.37). На основе полученных результатов мы пришли к выводу о преимуществе реализованных в PASS дескрипторов MNA и алгоритма классификации, по сравнению с используемыми в SuperPred и SwissTargetPrediction методах оценки по сходству или поиску ассоциаций на основе молекулярного докинга в DRAR-CPI [49]. В 2017 году было сопоставлено качество прогноза исходных и репозиционированных фармакотерапевтических эффектов с использованием шести доступных в Интернете веб ресурсов (ChemProt, PASS, SEA, SuperPred, SwissTargetPrediction, TargetHunter) с использованием двух тестовых выборок: 50 репозиционированных лекарств и 12 препаратов, недавно запатентованных по новому назначению [50]. Для первой выборки значения чувствительности варьировали от 0.64 (TarPred) до 1.00 (PASS) для исходных показаний, и от 0.64 (TarPred) до 0.98 (PASS) для репозиционированных показаний. Для второй выборки - от 0.08 (SuperPred) до 1.00 (PASS) для исходных показаний, и от 0.00 (SuperPred) до 1.00 (PASS) для репозиционированных показаний. Таким образом был сделан вывод о «самодостаточности» прогноза PASS и отсутствии необходимости консенсусных прогнозов на основе комбинирования результатов PASS и каких-либо других веб ресурсов [50]. ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ ПРОГНОЗА PASS Пользователь PASS получает результат прогноза спектра биологической активности в виде упорядоченного списка оценок вероятностей Pa и Pi принадлежности прогнозируемого соединения к классам «активных» и всех прочих соединений, и названий соответствующих активностей. Упорядочение выполняется по убыванию разности Pa-Pi, так что более вероятные виды активности находятся в верхней части спрогнозированного спектра. Спрогнозированный спектр активности может анализироваться любым желаемым образом, но по умолчанию в него включаются активности, для которых Pa > Pi. Необходимо помнить, что вероятность Pa отражает, прежде всего, сходство структуры молекул данного органического соединения со структурами молекул, наиболее типичных в соответствующем подмножестве «активных» соединений в обучающей выборке. Поэтому никакой прямой корреляции вычисляемых величин Pa с количественными характеристиками активности, как правило, нет. Действительно активное соединение, но имеющее нетипичную для обучающей выборки структуру молекул, может иметь согласно прогнозу низкое значение Pa, даже, возможно, Pa < Pi, поскольку значения величин Pa для активных и Pi для неактивных соединений из обучающей выборки (подсчитанные с их исключением!) распределены строго равномерно, что следует из способа построения функций Pa(B) и Pi(B) [38]. Необходимо также помнить о том, что основное для алгоритма PASS выражение (9b) можно записать в аналогичном (4) виде:
Другой важный аспект интерпретации результатов прогноза связан с новизной анализируемого соединения по сравнению с соединениями в обучающей выборке. Результатам прогноза 0.3 < Pa < 0.7 примерно соответствуют наиболее вероятные структуры активных соединений в SAR Base (наибольший наклон на рисунке 4), хотя в силу (10a), оно и может сильно отличаться от всех соединений с данной активностью в SAR Base, но, вероятнее всего, оно «такое же». Если же Pa > 0.7, то шансы обнаружить активность в эксперименте довольно высоки, и соединение, скорее всего, сочетает в себе наиболее важные особенности активных соединений, имеет очень мало общего с остальными соединениями в SAR Base (левый нижний угол на рисунке 5) и даже может оказаться родоначальником нового химического класса для рассматриваемого вида биологической активности. Еще один важный аспект интерпретации результатов прогноза состоит в предположении, что характеристики выборки соединений, для которых выполнен прогноз, подобны характеристикам соединений в SAR Base, для которых построены оценки Pa и Pi, что необходимо для применимости имеющихся оценок Pa и Pi для анализа прогноза. Необходимо помнить, что в обычных выборках очень мало активных соединений. Например, в SAR Base PASS (версия 2017) для половины из 5050 прогнозируемых активностей имеется менее 30 активных соединений (в среднем 473). Таким образом, для 2525 видов активности априорная вероятность P(A) менее 0.00003, и в среднем по всем активностям P(A)=0.00046. Даже для наиболее «популярных» видов активности она мала -- для «Beta-lactamase AmpC inhibitor» (см. рис. 1) P(A)=0.02. Эти оценки означают, что в чисто случайной выборке из тысячи соединений априорная вероятность найти хотя бы одно соединение с заданной активностью менее 1/2. Поскольку в SAR Base PASS включаются только те соединения, для которых известна хотя бы одна найденная экспериментально активность, то в общем случае активные соединения встречаются еще реже. Если в исследуемой выборке N1 соединений с желаемой активностью и N0 неактивных соединений, то по результатам прогноза PASS при заданном пороге Pa условно активными будет признано N1(1-Pa)+N0Pi соединений, среди которых действительно активных всего N1(1-Pa) соединений. В силу редкой встречаемости активных соединений даже при высокой точности прогноза вполне возможно, что N1(1-Pa) ≪ N0Pi - ложноположительных прогнозов гораздо больше, чем истинных. Совершенно аналогично в SAR Base PASS (версия 2017) в среднем одно соединение имеет в спектре биологической активности менее трех видов активности, хотя, например, для Топирамата их 239. При прогнозе из 5050 прогнозируемых видов активности будет около сотни активностей с Pa>0.5. Описанный выше избыток ложноположительных предсказаний -- следствие редкой удачи найти активное соединение, однако использование прогноза PASS может в десятки раз сократить объем необходимого экспериментального тестирования по сравнению со слепым поиском. Обширный спрогнозированный спектр активности свидетельствует о том, что структура молекулы данного органического соединения довольно проста, не содержит каких-либо особенностей, обеспечивающих высокую селективность его биологического действия. Например, если дескрипторов MNA менее 20, то при пороге Pa>Pi результат прогноза PASS (версия 2017) может содержать более тысячи видов активности, тогда как если дескрипторов MNA более 40, то, как правило, он будет включать менее двухсот активностей. Eсли при прогнозе оказалось, что в структуре есть несколько новых по отношению к составу обучающей выборки дескрипторов MNA, то структура менее похожа на любую из структур в SAR Base, и результаты прогноза необходимо рассматривать как приблизительные оценки. При анализе прогнозируемых PASS спектров биологической активности необходимо учитывать реальные возможности экспериментального тестирования. При этом общей рекомендацией является последовательное исследование различных прогнозируемых видов биологической активности, от наиболее вероятных к менее вероятным. В таблице приведен пример прогноза спектра биологической активности для лекарственного препарата Топирамат (Topiramate).
Согласно сведениям, содержащимся в базе данных Integrity [51], Топирамат имеет следующие фармакотерапевтические показания: Treatment of Bipolar Disorder, Psychiatric Disorders (Not Specified), Treatment of Alcohol Dependency, Prophylactic Treatment of Migraine, Agents for Inflammatory Bowel Disease, Antiobesity Drugs, Antimigraine Drugs, Treatment of Cocaine Dependency, Treatment of Neuropathic Pain, Antiepileptic Drugs, Treatment of Substance Dependency, Treatment of Eating Disorders, Aid to Smoking Cessation, Treatment of Nutritional Disorders, Treatment of Cerebrovascular Diseases, Treatment of Obsessive-Compulsive Disorder (OCD). Как видно из приведенных на рисунке 6 результатов прогноза, большая часть этих эффектов успешно прогнозируется PASS. В базе данных Integrity содержится информация о взаимодействии Топирамата с ферментами метаболизма лекарств CYP3A4 и CYP2C19, что также нашло свое отражение в результатах прогноза. В Integrity приведена информация о следующих молекулярных механизмах действия Топирамата: Sodium Channel Blockers, Carbonic Anhydrase Type II Inhibitors, AMPA Antagonists, Kainate Antagonists. Блокирование Топираматом натриевых каналов прогнозируется с вероятностью Pa=0.710. Прогноз указывает на возможность взаимодействия Топирамата с карбоангидразой II (Carbonic anhydrase II stimulant, Pa=0.988; Carbonic anhydrase II inhibitor, Pa=0.563), однако не позволяет прийти к заключению о направлении воздействия (стимуляция или ингибирование). В таких случаях, когда одновременно прогнозируется агонистическое или антагонистическое действие на рецепторы, стимуляция или ингибирование ферментов, открытие или блокада каналов, и т.п., требуется детальное изучение характера воздействия в зависимости от дозы (известны ситуации, когда лекарственное вещество проявляет противоположные эффекты при разных дозах). Возможно, именно этим обстоятельством объясняется наличие в прогнозе антиглаукомного действия препарата, одним из известных механизмов которого является ингибирование карбоангидразы II. В то же время установлено, что у отдельных пациентов применение Топирамата приводит к возникновению глаукомы [52]. В прогнозе побочных эффектов PASS Online также имеется указание на возможность возникновения глаукомы. Такие молекулярные механизмы действия Топирамата, как AMPA и Kainate Antagonists PASS не прогнозирует, что указывает на невысокое структурное сходство данного препарата с наиболее типичными молекулами, имеющими эти виды активности, в обучающей выборке PASS. Для Топирамата также прогнозируется взаимодействие с ГАМК рецепторами, не указанное в Integrity [51], однако в литературе имеются указания на наличие такой биологической активности у препарата [53]. Для прогнозируемого эффекта Топирамата на имидазолиновые рецепторы (Imidazoline I1 receptor agonist), и антиаллергического действия (Antiallergic) нам не удалось найти экспериментального подтверждения в литературе. Эти виды биологической активности, наряду с возможным действием на другие формы карбоангидразы (Carbonic anhydrase V inhibitor, Carbonic anhydrase IX inhibitor, Carbonic anhydrase I inhibitor), целесообразно протестировать в эксперименте. Перечень прогнозируемых PASS видов биологической активности включает в себя как фармакотерапевтические эффекты, так и механизмы действия, что существенным образом отличает данный подход от других упомянутых выше методов (SEA, TarPred и др.), которые, в основном, предсказывают действие вещества на молекулярные мишени. Это позволяет использовать PASS для решения различных задач при поиске и создании новых лекарственных препаратов:
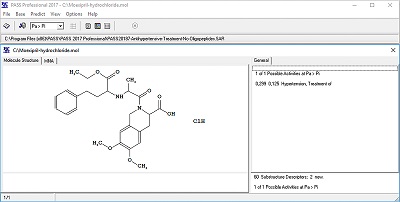
Рассмотрим некоторые примеры практического применения программы PASS в поиске и разработке новых фармакологических веществ. ПРИМЕРЫ ПРИМЕНЕНИЯ PASS Отбор на основе прогноза PASS наиболее перспективных соединений для синтеза и биологического тестирования. В рамках международного проекта INTAS был выполнен прогноз анксиолитического действия 5494 виртуальных структур из различных химических классов (тиазолы, пиразолы, изатины, имидазолы и др.), на основе которого было отобрано 8 наиболее перспективных соединений для синтеза и тестирования целевой активности [54]. Соединения были синтезированы и исследованы по стандартным фармакологическим тестам на лабораторных животных. Шесть из восьми исследованных соединений проявили анксиолитическую активность на уровне или выше препарата сравнения Медазепам. Структурные формулы пяти исследованных соединений существенно отличались от структуры известных анксиолитиков, что позволило отнести их к классу NCE (New Chemical Entities), то есть соединений, относящихся к химическим классам, в которых анксиолитическая активность ранее не была установлена [54]. С использованием прогноза PASS на основе виртуального скрининга химической библиотеки, содержащей 2648 органических молекул, было отобрано 32 «хита», для которых прогнозировалось ингибирование ксантиноксидазы, как потенциальных препаратов для лечения гиперурикемии [55]. 24 соединения были доступны в виде синтезированных образцов, и для них был проведен молекулярный докинг с помощью программы Glide XP (Schrödinger) по отношению к центру взаимодействия ксантиноксидазы с пираксостатом (идентификатор 3D структуры в PDB - 1VDV). Все 24 соединения были протестированы in vitro; обнаружено 3 ингибитора ксантиноксидазы; наиболее активное соединение имело значение IC50=1.4 мкM (для препарата сравнения, Аллопуринола, значение IC50=5.7 мкM). Анти-гиперурикемическое действие найденных соединений было подтверждено в экспериментах in vivo на крысах. Таким образом, авторы существенно сузили «пространство поиска» и докировали менее 1% соединений из всей химической библиотеки, отобранных на основе прогноза PASS. В конечном итоге, вместо полного скрининга всей библиотеки из 2648 соединений было протестировано 24 молекулы и найдено 3 активных вещества, то есть вероятность выявления активного соединения составила 12.5%, в то время как при случайном скрининге она составляет менее 1% [56]. Установление наиболее релевантных тестов для конкретных соединений. Несколько примеров практического применения PASS приведено в публикации Е.В. Бабаева [57]. Так, для новых производных 1-амино-4-(1,3-азолил-2)бутадиенов-1,3 наиболее высокой была вероятность противомикробной активности, причем прогноз был однотипен для всех синтезированных веществ. Антимикробное действие было подтверждено в экспериментах на грамположительных (Staphylococcus Aureus) и грамотрицательных (Escherichia coli) микроорганизмах. В ходе изучения новых химических превращений были получены ранее неизвестные индолизины с донорными заместителями. Хотя по своей структуре полученные соединения напоминали психотропные индолы псилоцинового ряда, для некоторых молекул PASS прогнозирует с высокой вероятностью связывание с бета-2 адренорецепторами. Прогнозируемая активность была подтверждена в эксперименте на мембранах синаптосом мозга крыс. Согласно прогнозу PASS, синтезированные по оригинальной методике новые производные имидазолов могли обладать антипротозойной активностью и быть эффективны против тропической лихорадки — лейшманиоза. Тестирование антилейшманиозной активности, проведенное в Университете г. Карачи (Пакистан), показало, что активность полученных соединений не уступает антилейшманиозному действию стандартного препарата Амфотерицина, недостатком которого является высокая токсичность [57]. Таким образом, приведенные выше примеры убедительно демонстрируют, каким образом предсказания PASS могут быть использованы для выбора наиболее перспективных тестов для изучения биологической активности конкретных соединений. Выявление новых соединений с требуемым набором видов биологической активности. С целью поиска новых антигипертензивных веществ, обладающих дуальными механизмами действия, c использованием PASS было выполнено прогнозирование ассоциированных с этим эффектом 30 молекулярных механизмов действия для 183462 молекул из баз данных компаний AsInEx и ChemBridge [58]. Тестирование in vitro четырех соединений, для которых было предсказано ингибирование ангиотензинконвертирующего фермента (ACE) и нейтральной эндопептидазы (NEP), подтвердило наличие у них прогнозируемых видов активности (для ACE значения IC50 = 10-7 - 10-9 M, для NEP значения IC50 = 10-5 M). Также на основе прогноза были выявлены вещества с такими дуальными механизмами действия, комбинации которых ранее не были описаны в литературе. На основе компьютерного прогноза ингибирования циклооксигеназ (COX) и липоксигеназы (LOX) для 573 виртуальных структур были отобраны для синтеза и биологического тестирования соединения, согласно прогнозу обладающие дуальными механизмами противовоспалительного действия [59]. Для синтеза и экспериментального тестирования было отобрано 9 производных 2-(тиазол-2-иламино)-5-фенилиден-4-тиазолидинона. Восемь изученных соединений проявили активность на широко используемой модели карагинанового воспаления; при этом 7 соединений ингибировали LOX, 7 соединений ингибировали COX, и 6 соединений ингибировали оба фермента [59]. Таким образом, продемонстрировано, что на основе анализа прогнозируемых с помощью PASS спектров биологической активности можно отбирать соединения, обладающие целевыми комбинациями механизмов действия и требуемыми фармакологическими эффектами. Выявление новых эффектов или механизмов действия для известных веществ. Компанией «Oriflame Skin Research Institute» (Швеция) было показано, что ацетил аспарагиновая кислота (AAA) проявляет омолаживающие кожу свойства (anti-ageing action), однако механизм этого действия оставался неясным [60]. Прогноз спектра биологической активности с использованием PASS показал, что AAA может стимулировать регенерацию кератиноцитов благодаря ингибированию действия матричных металлопротеиназ 1-3 и экспрессии F-актина. Проведенные in vitro исследования полностью подтвердили результаты прогноза PASS, несмотря на то, что указанные выше виды биологической активности прогнозировались с невысокими вероятностями. Прогноз спектров биологической активности для разрешенных к медицинскому применению лекарственных препаратов позволил предположить, что ряд антигипертензивных средств, ингибиторов ангиотензинконвертирующего фермента, включая Каптоприл, Эналаприл, Рамиприл и др., может обладать ноотропным действием [61]. Ноотропную активность трех препаратов этого класса исследовали на мышах по тесту спонтанной ориентации (поведения патрулирования) в крестообразном лабиринте. Было показано, что Периндоприл в дозе 1 мг/кг, а Квинаприл и Моноприл в дозе 10 мг/кг вызывают улучшение показателей поведения патрулирования лабиринта, сходным образом с эффектами референсных ноотропных препаратов Пирацетама и Меклофеноксата (в дозах 300 и 120 мг/кг соответственно). Установленное ноотропное действие некоторых ингибиторов АПФ, скорее всего, не связано с их антигипертензивным эффектом, поскольку ноотропное действие имело место лишь при относительно малых дозах периндоприла, квинаприла и моноприла, и исчезало при дальнейшем увеличении дозы. Выявление ноотропных свойств у антигипертензивных препаратов открывает возможности для их нового применения в медицинской практике, что было впоследствии подтверждено в клинике [62]. Обзор некоторых публикаций с результатами прогнозов биологической активности, полученных с использованием программы PASS, приведен в работах [36, 37]. ОТВЕТЫ НА ЧАСТО ЗАДАВАЕМЫЕ ВОПРОСЫ ПОЛЬЗОВАТЕЛЕЙ PASS Выбор пороговых значений для дифференциации активных и неактивных молекул. В разделе про интерпретацию результатов прогноза PASS подчеркнуто, что в SAR Base PASS (версия 2017) для половины активностей априорная вероятность P(A) менее 0.00003 и 0.00046 в среднем по всем активностям. Это может служить ориентиром ожидаемого успеха при слепом поиске активных соединений – необходимо выполнить биологические испытания до десятков тысяч соединений для обнаружения хотя бы одного активного соединения. Использование прогноза PASS может сократить необходимые объемы экспериментальных исследований в десятки раз. Но выбрать одно какое-то пороговое значение Pa или Pi невозможно – мы рекомендуем следовать стратегии последовательного испытания соединений (активностей) в порядке убывания значений Pa (Pa-Pi) в результатах прогноза, при таком подходе будет самой высокой вероятность достижения первого успеха. Выше мы уже отмечали, что вероятность наличия активности Pa отражает сходство структуры молекул анализируемого вещества со структурами молекул «наиболее типичных» в соответствующем подмножестве активных соединений в обучающей выборке. Несмотря на то, что мы стараемся поддерживать актуальное состояние обучающей выборки, постоянно проводя поиск новой и уточнение имеющейся в обучающей выборке информации, очевидно, что размерность пространства известных биологически активных соединений существенно уступает размерности теоретически возможных органических молекул. Чтобы проиллюстрировать, каким образом недостаток в обучающей выборке информации о конкретных химических классах соединений может повлиять на качество прогноза, были проведены специальные компьютерные эксперименты. Для этого мы извлекли из базы данных Integrity [51] данные о соединениях, удовлетворяющих запросу: “Hypertension treatment” {Therapeutic Group} AND “Chemical Categories” {Product Category} AND “To 1250” {Molecular Weight}. Таких документов оказалось 1924. После фильтрации с помощью разработанной нами процедуры “CheckSDF” на основе вышеописанных критериев PASS для структур соединений, их осталось 1762: 162 документа было отсеяно, поскольку 112 записей содержали два и более компонентов; 45 записей содержали заряженные молекулы; 4 записи содержали молекулы, у которых было более разрешенного числа аминокислотных остатков; 1 запись содержала символ атома, не соответствующий таблице Менделеева. После проведения процедуры обучения программы PASS в базе знаний SAR Base оказалось 1655 молекул, имеющих 3967 разных дескрипторов MNA 1-го и 2-го уровней, с 809 наименованиями биологической активности. После проведения селекции мы оставили 78 видов активности, ассоциированных с антигипертензивным действием; при этом среднее качество прогноза по критерию IAP, оцененное по процедуре скользящего контроля с исключением по одному, составило 0.9840. Точность прогноза для отдельных видов активности варьировала от 0.8898 до 1.0000. В процедуре скользящего контроля со случайным разбиением выборки на 20 частей среднее по активностям значение IAP равно 0.9841, что свидетельствует о достаточно высокой точности и предсказательной способности полученной нами SAR Base. В дальнейшем мы сконцентрировались на единственной активности “Antihypertensive”. С помощью имеющихся в Integrity возможностей информационного поиска мы проанализировали, какие химические классы (Product Category) составляют полученную нами выборку. Оказалось, что около половины молекул относятся к классу “Oligopeptides, less than 10 AA” (олигопептиды, содержащие менее 10 аминокислотных остатков). Далее мы разбили эту обучающую выборку на две подвыборки: первая включала в себя все молекулы, не отнесенные к классу “Oligopeptides, less than 10 AA” и была использована в качестве обучающей, а вторая, содержащая такие олигопептиды, была использована в качестве тестовой выборки. После обучения оказалось, что в SAR Base включено 792 молекулы, и для антигипертензивной активности IAP=0.8791. Прогноз антигипертензивной активности на основе исходной обучающей выборки, содержащей все извлеченные из Integrity молекулы, при пороге Pa>Pi позволяет правильно классифицировать 889 из 939 молекул (~95%). На основе новой обучающей выборки, не содержащей молекул олигопептидов, прогноз антигипертензивной активности при пороге Pa>Pi позволяет правильно классифицировать 752 из 939 молекул (~80%). Среднее значение Pa для предсказанных активными молекул в первом случае составило 0.675, в то время как во втором случае это значение снизилось до 0.394. Еще более разительные отличия наблюдались при исключении из обучающей выборки веществ, отнесенных к категории “Nucleosides”. В этом случае после обучения SAR Base содержала 1508 молекул, а для антигипертензивной активности IAP=0.9354. Прогноз антигипертензивной активности на основе исходной обучающей выборки, содержащей все извлеченные из Integrity молекулы, при пороге Pa>Pi позволяет правильно классифицировать 136 из 166 молекул (~82%). Прогноз антигипертензивной активности на основе новой обучающей выборки, не содержащей молекулы нуклеозидов, при пороге Pa>Pi позволяет правильно классифицировать 89 из 166 молекул (~54%). При этом среднее значение Pa для предсказанных активными молекул в первом случае составило 0.332, в то время как во втором случае это значение снизилось до 0.245. Полученные в ходе этих компьютерных экспериментов результаты указывают на существенную зависимость получаемых при прогнозе оценок значений Pa от того, насколько структура анализируемого соединения сходна со структурой включенных в обучающую выборку веществ, имеющих конкретный вид активности. Поскольку PASS прогнозирует несколько тысяч видов биологической активности и для каждого из них величины сходства анализируемого соединения с соответствующими соединениями обучающей выборки могут отличаться, нельзя предложить единый универсальный критерий для выбора «порога», позволяющего по результатам прогноза PASS отделить «активные» соединения от «неактивных». Именно поэтому «по умолчанию» в PASS принято пороговое значение Pa>Pi, особенности которого можно видеть на рисунке 5. Мы рекомендуем использовать результат прогноза для референсного соединения (применяемого в фармакологических исследованиях препарата сравнения) как реперной точки. Так, например, в случае антигипертензивных препаратов олигопептидной природы референсным соединением может быть разрешенный к медицинскому применению препарат Moexipril Hydrochloride [51]. Как видно из результата прогноза PASS на рисунке 6, при использовании SAR Base, не содержащей олигопептидов, для этой молекулы значение Pa равно 0.299. Если выбрать это значение в качестве порогового, то при использовании данной SAR Base из 752 соединений, для которых Pa>Pi, можно будет выявить 466 с Pa>0.299.
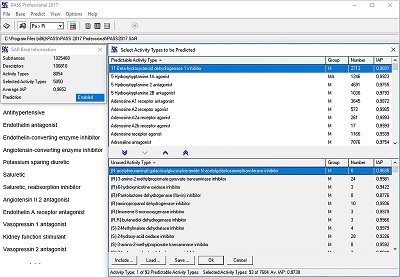
Дополнительные погрешности при рассмотрении прогностических оценок могут вносить так называемые “Activity cliffs”, когда сравнительно небольшие изменения структуры приводят к резкому падению (либо возрастанию) величины активности [63, 64]. Очевидно, что существующие методы анализа зависимостей «структура-активность», как правило, основаны на предположении о существовании «гладких зависимостей», что не позволяет в большинстве случаев идентифицировать такого рода ситуации. В этой связи, даже если в прогнозе нет интересующей пользователя биологической активности, но он по каким-либо причинам уверен, что такая активность должна присутствовать, имеет смысл проверить экспериментально ее наличие. Ограничение перечня прогнозируемых видов активности. В программе PASS реализована процедура селекции интересующих пользователя видов биологической активности еще до проведения прогноза. Это дает возможность снизить временные затраты на анализ результатов прогноза, заранее ограничив их либо интересующей фармакотерапевтической областью, либо теми видами активности, для которых пользователь может организовать проведение экспериментального тестирования. Выбор интересующих видов активности может быть выполнен либо вручную, по одному, либо путем загрузки списка целевых видов активности. Так, например, на рисунке 7 представлена загрузка списка видов активности, ассоциированных с антигипертензивным действием.
В результате, вместо 5050 видов биологической активности, прогнозируемых по умолчанию PASS (2017) со средней точностью IAP = 0.9652, пользователь получит прогноз 93 интересующих его видов активности с чуть более высокой средней точностью IAP = 0.9738. Границы применимости прогноза PASS. Возможности PASS ограничены перечнем прогнозируемых видов активности, который составлен с учетом современного состояния фармакологической науки, и источниками данных о результатах экспериментального исследования органических соединений на биологическую активность. Создание новых биологически активных веществ – динамично развивающаяся область, постоянно претерпевающая как количественные, так и качественные изменения. Появляются новые термины, характеризующие биологическую активность, например, “Drugs Modulating Gene Expression”, “Transcription Factor Ligands”, “Translation Initiation Factor Inhibitors” [51] или “Modulators of Alternative Splicing” [65], и др. Усложняется наше понимание связей между молекулярными механизмами действия и вызываемыми ими фармакотерапевтическими эффектами, что привело к появлению и развитию «сетевой фармакологии» [66]. Соответственно, необходимы постоянные усилия, направленные не только на то, чтобы пополнять обучающую выборку PASS новыми данными о структуре и биологической активности органических соединений, но и существенным образом уточнять понятийный аппарат описания химико-биологических взаимодействий в ряду «лиганд - мишень – биологический процесс – болезнь» [67]. Кроме того необходимо отметить, что критерии отнесения соединений к «активным» и «неактивным» также изменяются со временем. В случае исследования новых мишеней, для которых лиганды либо не известны, либо их активность сравнительно невысока, соединения со значениями IC50 < 100 мкМ могут рассматриваться как активные. Если для рассматриваемой мишени уже известны вещества, действующие в микромолярных или даже субмикромолярных концентрациях, этот порог снижается до IC50 < 10 мкМ, или даже до IC50 < 1 мкМ. Сложности интерпретации количественных данных об активности также связаны с различием методик экспериментального тестирования [68-70]. Отчасти вносимые этими факторами погрешности компенсируются статистической устойчивостью используемого в PASS подхода [38]. Необходимо помнить, что PASS прогнозирует возможность проявления биологической активности конкретным соединением, однако не позволяет сделать каких-либо умозаключений относительно величины активности и условий экспериментального тестирования (доза, путь введения, биологический объект, пол, возраст и т.п.), при которых эта активность может проявиться. Таким образом, PASS позволяет сузить область экспериментального тестирования в отношении конкретных соединений, однако любой прогноз необходимо подтверждать экспериментом. Особую осторожность следует проявлять при интерпретации прогнозируемых PASS побочных или токсических эффектов, поскольку эти эффекты могут не только проявляться при существенно более высоких, в сравнении с терапевтическими, дозах, но также могут наблюдаться у сравнительно небольших групп пациентов (известно, что многие побочные эффекты возникают вследствие идиосинкратических реакций на прием лекарств) [71]. Следует также подчеркнуть, что PASS не может предсказать, станет ли конкретное вещество лекарственным препаратом, поскольку это зависит от ряда различных факторов. Предсказание, однако, может помочь определить, на какие виды биологической активности следует протестировать анализируемое соединение в первую очередь, и какие вещества с наибольшей вероятностью могут проявить требуемые виды активности. Необходимость нормализации структуры молекул до выполнения прогноза. Как отмечалось выше, во всех современных исследованиях, направленных на компьютерный анализ зависимостей «структура-активность», необходимо предварительно «нормализовать структуру» (убрать солевой компонент, нейтрализовать заряды, заменить координационные связи простыми и т.д.) [42-44]. Одной из широко используемых для нормализации структуры молекул является компьютерная программа Standardizer фирмы ChemAxon [72]. Не все применяемые при этом операции интуитивно понятны химикам-синтетикам, однако их выполнение необходимо для того, чтобы обеспечить однородность представления структурной химической информации как в обучающей выборке PASS, так и в структурах, направляемых на прогноз их биологической активности. Как уже говорилось выше, в PASS имеются некоторые дополнительные ограничения на анализируемые структуры: наличие не менее трех атомов углерода и молекулярный вес, не превосходящий 1250 a.e.m. Эти ограничения связаны с необходимостью обеспечить соответствие между молекулами, направляемыми на прогноз, и молекулами, содержащимися в обучающей выборке (прогноз должен выполняться для молекул, попадающих в область применимости зависимостей «структура-активность», представленных в SAR Base). ПРИМЕРЫ НЕТОЧНОСТЕЙ ПРИ ИНТЕРПРЕТАЦИИ ПРОГНОЗА PASS Отсутствие экспериментального подтверждения прогноза. Наиболее типичной ситуацией, с которой приходится сталкиваться при рассмотрении некоторых опубликованных работ, является отсутствие экспериментального подтверждения прогнозируемых PASS видов биологической активности. Это нельзя назвать «ошибкой», поскольку, по-видимому, у исследователя просто нет возможности проведения соответствующих экспериментальных исследований. В то же время, ошибочным является утверждение, что вещества, для которых получен прогноз, обладают прогнозируемыми видами активности. Так, например, в работе [73] осуществлен синтез N-(2,5-диметил-4-нитрофенил)-4-метилбензолсульфонамида (NDMPMBS), для которого выполнен прогноз спектра биологической активности с использованием PASS. Ряд активностей прогнозируется с достаточно высокой вероятностью, включая Arylsulfate sulfotransferase inhibitor (Pa=0.889), Polyporopepsin inhibitor (Pa=0.888), Glutamyl endopeptidase II inhibitor (Pa=0.860), Phospholipid-translocating ATPase inhibitor (Pa=0.850), и др. Авторы делают вывод о перспективности синтезированного вещества для использования в фармацевтических целях («Results provided … indicate great potential of the newly synthesized NDMPMBS molecule for application in pharmaceutical applications»). Очевидно, что это – слишком оптимистичное утверждение, которое не обосновано полученными в цитируемой работе результатами. «Подтверждение» прогноза PASS на основе молекулярного докинга. Иногда встречаются публикации, в которых для некоторых молекулярных мишеней, взаимодействие с которыми прогнозируется PASS, проводится молекулярный докинг, по результатам которого утверждается, что изучаемые соединения обладают конкретным видом биологической активности. Так, например, в работе [74], авторы выполнили с помощью PASS прогноз спектров биологической активности для некоторых алкалоидов из растений рода Strychnos. Одним из прогнозируемых для стризонобразилина (Strychnobrasiline) видов активности является противоопухолевое действие (Pa=0.396). Используя AutoDock-Vina, авторы выполнили докинг этого вещества к комплексу ДНК с топоизомеразой II, проанализировали возможности взаимодействия стризонобразилина с этой мишенью. Аналогичный докинг был выполнен для 12-гидрокси-10,11-стризонобразилина, в результате чего авторы пришли к заключению о более высокой перспективности этого производного по сравнению с исходной молекулой (интересно, что для 12-гидрокси-10,11-стризонобразилина противоопухолевое действие прогнозируется с более высокой вероятностью Pa=0.622). В то же время, эти умозаключения требуют экспериментальной проверки, которая в цитируемой работе [74] не была осуществлена. Интересно, что схожие выводы были сделаны авторами работы [75], которые на основе прогноза с помощью Swiss Target Prediction выбрали в качестве молекулярной мишени серотониновый транспортер. Докинг с использованием AutoDock 4.0, по мнению авторов цитируемой работы, позволяет прийти к заключению, что молекула 3-(1,8-дихлоро-9,10-дигидро-9,10-этаноантрацен-11-yl)акрилальдегида является эффективным антидепрессантом («Docking study indicated that compound 2 is a good antidepressant-like compound»). Понятно, что это умозаключение не вполне корректно, учитывая известные ограничения методов докинга [13]. Рассмотрение полного спектра биологической активности в качестве «подтверждения» перспективности изучаемых веществ. Как было указано выше, для «простых» веществ, не имеющих существенных структурных особенностей, прогнозируемый спектр биологической активности может оказаться чрезвычайно широким. Так, например, в работе [76] приведен прогнозируемый спектр биологической активности для бета-элемена, включающий 629 видов биологической активности (последний в списке – Retinoic acid receptor antagonist, для которого значение Pa=0.022). Несмотря на то, что для отдельных предсказанных видов активности автор приводит литературные данные, подтверждающие их наличие, вывод относительно перспективности дальнейшего исследования этого соединения в качестве фармакологического вещества может оказаться неоправданно оптимистичным, поскольку относительно простые молекулы, действительно, могут связываться со многими молекулярными мишенями в биологических системах, но с низкими значениями аффинности и специфичности [10]. Таким образом, при анализе результатов предсказаний PASS необходимо учитывать сложность понятия биологической активности и возможные неоднозначности установления зависимостей «структура-активность», что требует достаточно высокой квалификации исследователя, и, несомненно, верификации прогноза в эксперименте. ЗАКЛЮЧЕНИЕ В настоящей работе мы рассмотрели используемый в PASS подход к прогнозированию биологической активности, основанный на анализе информации около 1 млн органических соединений с установленной биологической активностью, и привели рекомендации по корректной интерпретацию результатов предсказаний. Необходимо подчеркнуть, что эти рекомендации применимы как к стандартной версии PASS, прогнозирующей несколько тысяч видов биологической активности [36, 77], так и к специализированным версиям программы: PASS Targets [78, 79], DIGEP Pred [80, 81], PASS CLC Pred [82-84], SMP [85, 86], SOMP [87, 88], RA [89, 90], MetaTox [91-93], ADVER-Pred [94, 95], ROSC-Pred [96, 97], KinScreen [98]. Также необходимо указать, что если полученные пользователем результаты прогноза представляются ему не соответствующими известным литературным (или персональным) данным, у него имеется возможность добавления веществ соответствующего химического класса к обучающей выборке PASS с использованием веб ресурса SAR Creator [99]. Поскольку периодически мы осуществляем обновление SAR Base, проводя заново процедуру обучения, в следующих версиях программы PASS прогностические возможности для соединений данного химического класса будут расширены. Если и после обновления SAR Base прогноз будет неудовлетворительным, у пользователя будет возможность построения (Q)SAR моделей на основе подготовленной им обучающей выборки с использованием специализированных программ, например, программы GUSAR, основанной на применении QNA дескрипторов и самосогласованной регрессии [100-110]. БЛАГОДАРНОСТИ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. Авторы выражают искреннюю признательность компании Clarivate Analytics за предоставление лицензии на доступ к базе данных Integrity и компании ChemAxon за предоставление лицензии на JChem. ЛИТЕРАТУРА
|