Computer-aided prediction of biological activity spectra for chemical compounds: opportunities and limitations
1Institute of Biomedical Chemistry, 8119121 Pogodinskaya Str., 10 bldg. 8, Moscow, Russia,*e-mail: vladimir.poroikov@ibmc.msk.ru 2Pirogov Russian National Research Medical University, 117997, Ostrovityanova Str., 1, Moscow, Russia.
Keywords: analysis of structure-activity relationships, biological activity spectra, computer-aided prediction, PASS, accuracy, predictivity, web-resource Way2Drug
DOI: 10.18097/BMCRM00004
An essential characteristic of chemical compounds is their biological activity since its presence can become the basis for the use of the substance for therapeutic purposes, or, on the contrary, limit the possibilities of its practical application due to the manifestation of side action and toxic effects. Computer assessment of the biological activity spectra makes it possible to determine the most promising directions for the study of the pharmacological action of particular substances, and to filter out potentially dangerous molecules at the early stages of research. For more than 25 years, we have been developing and improving the computer program PASS (Prediction of Activity Spectra for Substances), designed to predict the biological activity spectrum of substance based on the structural formula of its molecules. The prediction is carried out by the analysis of structure-activity relationships for the training set, which currently contains information on structures and known biological activities for more than one million molecules. The structure of the organic compound is represented in PASS using Multilevel Neighborhoods of Atoms descriptors; the activity prediction for new compounds is performed by the naive Bayes classifier and the structure-activity relationships determined by the analysis of the training set. We have created and improved both local versions of the PASS program and freely available web resources based on PASS (http://www.way2drug.com). They predict several thousand biological activities (pharmacological effects, molecular mechanisms of action, specific toxicity and adverse effects, interaction with the unwanted targets, metabolism and action on molecular transport), cytotoxicity for tumor and non-tumor cell lines, carcinogenicity, induced changes of gene expression profiles, metabolic sites of the major enzymes of the first and second phases of xenobiotics biotransformation, and belonging to substrates and/or metabolites of metabolic enzymes.
The web resource Way2Drug is used by over 19 000 researchers from more than 100 countries around the world, which allowed them to obtain over 600 000 predictions and publish about 500 papers describing the obtained results. The analysis of the published works shows that in some cases the interpretation of the prediction results presented by the authors of these publications requires an adjustment. In this work, we provide the theoretical basis and consider, on particular examples, the opportunities and limitations of computer-aided prediction of biological activity spectra.


|
Figure 1.
A cloudy representation of all biological activities predicted by PASS (version 2017). The font size is proportional to the number of compounds with the appropriate activity in the training set.
|
|
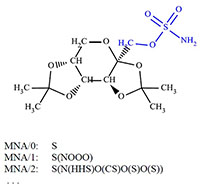
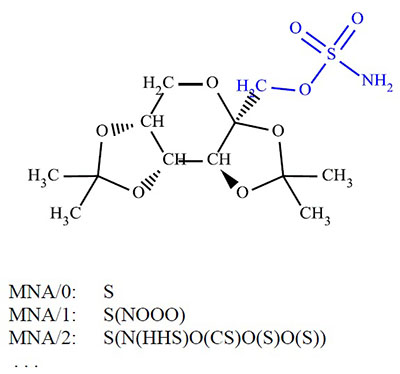
Figure 2.
MNA descriptors for the sulfur atom in the Topiramate molecule (PubChem CID: 5284627). The atoms of the fragment covered by MNA/2 descriptor is highlighted in blue.
|

|
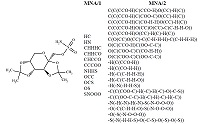
Figure 3.
The structural formula of the Topiramate molecule and the set of MNA descriptors describing the structure of this molecule in PASS.
|
|
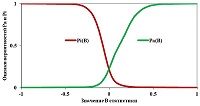
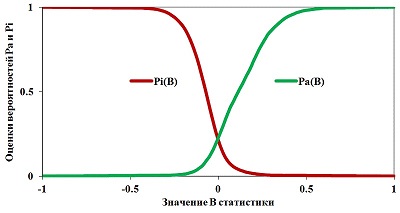
Figure 4.
Dependencies of Pa (B) and Pi (B) for "Antihypertensive" activity estimated using the data presented in SAR Base PASS version 2017.
|
|
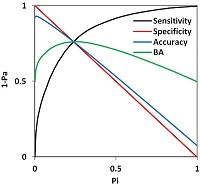
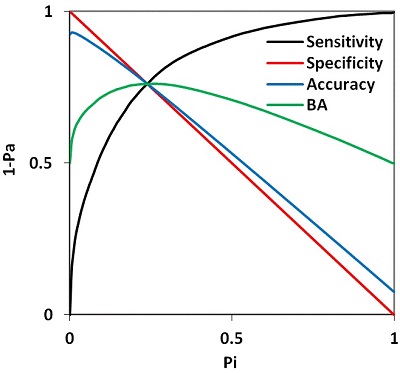
Figure 5.
An example of the relationships between the Sensitivity (Sensitivity ≡ 1-Pa, black curve), Specificity (Specificity ≡ 1-Pi, red line), Concordance (blue curv), and Balanced Accuracy (BA = (Sensitivity + Specificity) / 2) as a function of the threshold for the probability of errors of the second kind (Pi) for the activity "Antitumor"
|
|
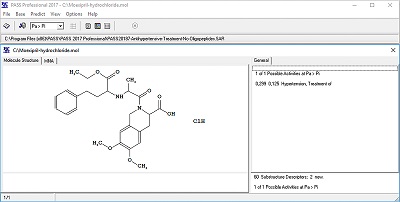
Figure 6.
An example of selecting threshold values Pa based on the prediction of antihypertensive activity for the reference drug Moexipril Hydrochloride.
|
|
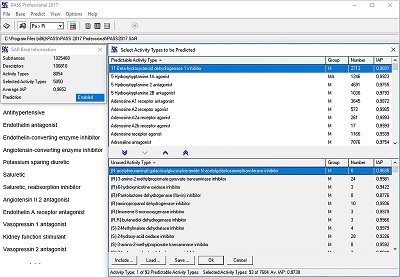
Figure 7.
Restricting the prediction of biological activity spectrum only by the list of mechanisms and effects associated with antihypertensive action.
|
|
CLOSE

|
Table.
Predicted by PASS online biological activity spectrum Topiramate molecule (PubChem CID: 5284627) at the threshold
|
ACKNOWLEDGEMENTS
The work was supported in the framework of the Russian State Academies of Sciences Fundamental Research Program for 2013-2020. The authors express sincere gratitude to the Clarivate analytics for the license on Integrity, and ChemAxon for the license on JChem.
REFERENCES
- Barenboim, G.M. & Malenkov, A.G. (1986) Biologically active substances. New techniques of discovery. Moscow: Nauka (Rus).
- Czerepak, E. & Ryser, S. (2008). Drug approvals and failures: implications for alliances. Nature Reviews Drug Discovery, 7, 197198. DOI
- ChemNavigator, Retrieved March 24, 2018, from http://www.chemnavigator.com/
- CAS. Retrieved March 24, 2018, from http://www.cas.org/
- SAVI. Retrieved March 24, 2018, from https://cactus.nci.nih.gov/download/savi_download/
- Ruddigkeit, L., Blum, L. C. & Reymond, J.-L. (2013). Visualization and virtual screening of the chemical universe database GDB-17. Journal of Chemical Information and Modeling, 53(1), 56-65. DOI
- Santos, R., Ursu, O., Gaulton, A., Bento, A. P., Donadi, R. S., Bologa, C. G., Karlsson, A., Al-Lazikani, B., Hersey, A., Oprea, T. I. & Overington, J. P. (2017). A comprehensive map of molecular drug' target=s. Nature Reviews Drug Discovery, 16(1), 19-34. DOI
- Li, Y. H., Yu, C. Y., Li, X. X., Zhang, P., Tang ,J., Yang, Q., Fu, T., Zhang, X., Cui, X., Tu, G., Zhang, Y., Li, S., Yang, F., Sun, Q., Qin, C., Zeng, X., Chen, Z., Chen, Y. Z. & Zhu, F. (2018). Therapeutic' target= database update 2018: enriched resource for facilitating bench-to-clinic research of' target=ed therapeutics. Nucleic Acids Research, 46(D1), D1121-D1127. DOI
- Chisholm-Burns, M. A., Schwinghammer, T. L., Wells, B. G., Malone, P. M., DiPiro, J. T., & Kolesar, J. M. (2015). Pharmacotherapy Principles and Practice, Fourth Edition. NY: McGraw Hill Professional.
- Lipinski, C. & Hopkins, A. (2004). Navigating chemical space for biology and medicine. Nature, 432, 855-861. DOI
- Ivanov, A.S., Poroikov, V.V., & Archakov, A.I. (2003) Bioinformatics: way from genome to drug in silico. Bulletin of RSMU, 4(30), 19-23 (Rus).
- Jorgensen, W. L. (2004). The many roles of computation in drug discovery. Science, 303(5665), 1813-1818. DOI
- Chen, Y. C. (2015). Beware of docking! Trends in Pharmacological Science, 36(2), 78-95. DOI
- Luo, H., Zhang, P., Cao, X. H., Du, D., Ye, H., Huang, H., Li, C., Qin, S., Wan, C., Shi, L., He, L. & Yang, L. (2016). DPDR-CPI, a server that predicts drug positioning and drug repositioning via chemical-protein interactome. Scientific Reports, 6, 35996. DOI
- Martin, Y. C., Kofron, J. L. & Traphagen, L. M. (2002). Do structurally similar molecules have similar biological activity? Journal of Medicinal Chemistry, 45(19), 4350-4358. DOI
- Bender, A. (2010). How similar are those molecules after all? Use two descriptors and you will have three different answers. Expert Opinion on Drug Discovery, 5(12), 1141-1151. DOI
- PubChem. Retrieved March 24, 2018, from https://pubchem.ncbi.nlm.nih.gov/
- ChEMBL. Retrieved March 24, 2018, from https://www.ebi.ac.uk/chembl/
- DrugBank. Retrieved March 24, 2018, from https://www.drugbank.ca/
- ChemProt. Retrieved March 24, 2018, from http://potentia.cbs.dtu.dk/ChemProt/
- SEA. Retrieved March 24, 2018, from http://sea.bkslab.org/
- SuperPred. Retrieved March 24, 2018, from http://prediction.charite.de/
- SwissTargetPrediction. Retrieved March 24, 2018, from http://www.swisstargetprediction.ch/
- TarPred. Retrieved March 24, 2018, from http://202.127.19.75:5555/
- TargetHunter. Retrieved March 24, 2018, from http://www.cbligand.org/TargetHunter/
- Mervin, L. H., Afzal, A. M., Drakakis, G., Lewis, R., Engkvist, O. & Bender, A. (2015).' target= prediction utilising negative bioactivity data covering large chemical space. Journal of Cheminformatics, 7, 51. DOI
- Burov, Yu.V., Poroikov, V.V., Korolchenko, L.V. (1990) National system for registration and biological testing of chemical compounds: facilities for new drugs search. Bulletin of National Center for Biologically Active Compounds, 1, 4-25 (Rus).
- Poroikov, V.V., Filimonov, D.A. & Boudunova, A.P. (1993) Comparison of the Results of Prediction of the Spectra of Biological Activity of Chemical Compounds by Experts and the PASS System. Automatic Documentation and Mathematical Linguistics, 27 (3), 40-43.
- Filimonov, D.A., Poroikov V.V., Karaicheva, E.I., Kazaryan, R.K., Boudunova, A.P., Mikhailovskiy, E.M., Rudnitskih, A.V., Goncharenko, L.V. & Burov, Yu.V. (1995) Computer-Aided Prediction of Biological Activity Spectra of Chemical Substances on the Basis of Their Structural Formulae: Computerized System PASS. Experimental and Clinical Pharmacology, 58(2), 56-62 (Rus).
- Filimonov, D. A. & Poroikov, V. V. (1996). PASS: computerized prediction of biological activity spectra for chemical substances. In Bioactive Compound Design: Possibilities for Industrial Use (pp. 47-56). Oxford, UK: BIOS Scientific Publishers.
- Poroikov, V.V. (1999) Computer-aided prediction of biological activity for chemical substances: possibilities and limitations. Chemistry in Russia, 2, 8-12 (Rus).
- Lagunin, A., Stepanchikova, A., Filimonov, D. & Poroikov, V. (2000). PASS: prediction of activity spectra for biologically active substances. Bioinformatics, 16(8), 747-748. DOI
- Poroikov, V. V., Filimonov, D. A., Ihlenfeldt, W.-D., Gloriozova, T. A., Lagunin, A. A., Borodina, Yu. V., Stepanchikova, A. V. & Nicklaus, M. C. (2003). PASS Biological Activity Spectrum Predictions in the Enhanced Open NCI Database Browser. Journal of Chemical Information and Computer Sciences, 43(1) 228-236. DOI
- Filimonov, D.A. & Poroikov, V.V. (2006) Prediction of biological activity spectra for organic compounds. Russian Chemical Journal, 50 (2), 66-75 (Rus)
- Filimonov, D. A. & Poroikov V. V. (2008). Probabilistic approach in activity prediction. In A. Varnek & A. Tropsha (Eds.) Chemoinformatics Approaches to Virtual Screening (pp. 182-216). Cambridge, UK: RSC Publishing.
- Filimonov, D. A., Lagunin, A. A., Gloriozova, T. A., Rudik, A. V., Druzhilovskiy, D. S., Pogodin, P. V. & Poroikov V. V. (2014). Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chemistry of Heterocyclic Compounds, 50(3), 444-457. DOI
- Druzhilovskiy, D. S., Rudik, A. V., Filimonov, D. A., Gloriozova, T. A., Lagunin, A. A., Dmitriev, A. V., Pogodin, P. V., Dubovskaja, V. I., Ivanov, S. M., Tarasova, O. A., Bezhentsev, V. M., Murtazalieva, K. A., Semin, M. I., Maiorov, I. S., Gaur, A. S., Sastry, G. N. & Poroikov, V. V. (2017). Computational platform Way2Drug: from the prediction of biological activity to drug repurposing. Russian Chemical Bulletin, International Edition, 66(10), 1832-1841. DOI
- Poroikov, V. V., Filimonov, D. A., Borodina, Yu. V., Lagunin, A. A. & Kos, A. (2000). Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. Journal of Chemical Information and Computer Sciences, 40(6), 1349-1355. DOI
- Filimonov, D., Poroikov, V., Borodina, Yu. & Gloriozova T. (1999). Chemical similarity assessment through multilevel neighborhoods of atoms: definition and comparison with the other descriptors. Journal of Chemical Information and Computer Sciences, 39(4), 666-670. DOI
- Holtje, H.-D., Folkers, G., Mannhold, R., Kubinyi, H. & Timmerman, H. (2008) Molecular modeling: Basic principles and applications. Weinheim: Wiley.
- Dalby, A., Nourse, G. J., Hounshell, W. D., Gushurst, A. K. I., Grier, D. L., Leland, B. A. & Laufer, J. (1992). Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. Journal of Chemical Information and Computer Science, 32(3), 244-255. DOI
- Fourches, D., Muratov, E. & Tropsha A. (2010). Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. Journal of Chemical Information and Modeling, 50(7), 1189-1204. DOI
- Fourches, D., Muratov, E. & Tropsha A. (2015). Curation of chemogenomics data. Nature Chemical Biology, 11(8), 535. DOI
- Fourches, D., Muratov, E. & Tropsha A. (2016). Trust, But Verify II: A Practical Guide to Chemogenomics Data Curation. Journal of Chemical Information and Modeling, 56(7), 1243-1252. DOI
- Townsend, J. A., Glen, R. C. & Mussa, H. Y. (2012). Note on naive Bayes based on binary descriptors in cheminformatics. Journal of Chemical Information and Modeling, 52(10), 2494-2500. DOI
- Mussa, H. Y., Mitchell, J. B. O. & Glen, R. C. (2013). Full УLaplacianisedФ posterior Naive Bayesian algorithm. Journal of Cheminformatics, 5, 37. DOI
- Mussa, H. Y., Marcus, D., Mitchell, J. B. & Glen, R. C. (2015). Verifying the fully "Laplacianised" posterior Naive Bayesian approach and more. Journal of Cheminformatics, 7, 27. DOI
- Geronikaki, A., Druzhilovsky, D., Zakharov, A. & Poroikov, V. (2008). Computer-aided predictions for medicinal chemistry via Internet. SAR and QSAR in Environmental Research, 19(1-2), 27-38. DOI
- Druzhilovskiy, D.S., Rudik, A.V., Filimonov, D.A., Lagunin, A.A., Gloriozova, T.A. & Poroikov V.V. (2016) Online resources for the prediction of biological activity of organic compounds. Russian Chemical Bulletin, International Edition, 65 (2), 384-393.
- Murtazalieva, K. A., Druzhilovskiy, D. S., Goel, R. K., Sastry, G. N. & Poroikov V. V. (2017). How good are publicly available web services that predict bioactivity profiles for drug repurposing? SAR and QSAR in Environmental Research, 28(10), 843-862. DOI
- Integrity. Retrieved March 24, 2018, from https://integrity.thomson-pharma.com/
- SleepDisorders. Retrieved March 24, 2018, from http://sleepdisorders.about.com/od/sleepdisorderstreatment/a/What-Is-Topamax.htm
- Bandini, F., Arena, E. & Mauro, G. (2012). Pre-orgasmic sexual headache responsive to topiramate: a case report. Cephalalgia, 32(10), 797-798. DOI
- Geronikaki, A., Babaev, E., Dearden, J., Dehaen, W., Filimonov, D., Galaeva, I., Krajneva, V., Lagunin, A., Macaev, F., Molodavkin, G., Poroikov, V., Saloutin, V., Stepanchikova, A. & Voronina, T. (2004). Design of new anxiolytics: from computer prediction to synthesis and biological evaluation. Bioorganic & Medicinal Chemistry, 12(24), 6559-6568. DOI
- B-Rao, C., Kulkarni-Almeida, A., Katkar, K. V., Khanna, S., Ghosh, U., Keche, A., Shah, P., Srivastava, A., Korde, V., Nemmani, K. V., Deshmukh, N. J., Dixit, A., Brahma, M. K., Bahirat, U., Doshi, L., Sharma, R. & Sivaramakrishnan H. (2012). Identification of novel isocytosine derivatives as xanthine oxidase inhibitors from a set of virtual screening hits. Bioorganic & Medicinal Chemistry, 20(9), 2930-2839. DOI
- Folmer, R. H. A. (2016). Integrating biophysics with HTS-driven drug discovery projects. Drug Discovery Today, 21(3), 491-498. DOI
- Babaev, E.V. (2009) Combinatorial chemistry in high school: ten-year experience of scientific, educational and organizational projects. Russian chemical journal, 53(5), 140-152 (Rus).
- Lagunin, A. A., Gomazkov, O. A., Filimonov, D. A., Gureeva, T. A., Dilakyan, E. A., Kugaevskaya, E. V., Elisseeva, Yu. E., Solovyeva, N. I. & Poroikov, V. V. Computer-aided selection of potential antihypertensive compounds with dual mechanisms of action. (2003). Journal of Medicinal Chemistry, 46(15), 3326-3332. DOI
- Geronikaki, A. A., Lagunin, A. A., Hadjipavlou-Litina, D. I., Elefteriou, P. T., Filimonov, D. A., Poroikov, V. V., Alam, I. & Saxena A.K. (2008). Computer-aided discovery of anti-inflammatory thiazolidinones with dual cyclooxygenase/lipoxygenase inhibition. Journal of Medicinal Chemistry, 51(6), 1601-1609. DOI
- Gillbro, J. M., Lundahl, M., Westman, M., Baral, R., Al-Bader, T. & Mavon A. (2015). Structural activity relationship analysis (SAR) and in vitro testing reveal the anti-ageing potential activity of acetyl aspartic acid. International Journal of Cosmetic Science, 37(S1), 15-20. DOI
- Kryzhanovskii, S.A., Salimov, R.M., Lagunin, A.A., Filimonov, D.A., Gloriozova, T.A. & Poroikov V.V. (2012) Nootropic action of some antihypertensive drugs: computer predicting and experimental testing. Pharmaceutical Chemistry Journal, 45 (10), 605-611.
- Gao, Y., OТCaoimh, R., Healy, L., Kerins, D. M., Eustace, J., Guyatt, G., Sammon, D. & Molloy, D. W. (2013). Effects of centrally acting ACE inhibitors on the rate of cognitive decline in dementia. BMJ Open, 3, 1-8. http://doi.org/10.1136/bmjopen-2013-002881
- Cruz-Monteagudo, M., Medina-Franco, J. L., Perez-Castillo, Y., Nicolotti, O., Cordeiro, M. N. & Borges, F. (2014). Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discovery Today, 19(8), 1069-1080. DOI
- Bajorath, J. (2017). Representation and identification of activity cliffs. Expert Opinion on Drug Discovery, 12(9), 879-883. DOI
- Bates, D. O., Morris, J. C., Oltean, S. & Donaldson, L. F. (2017). Pharmacology of modulators of alternative splicing. Pharmacological Reviews, 69(1), 63-79. DOI
- Hopkins, A. L. (2007). Network pharmacology. Nature Biotechnology, 25(10), 1110-1111. DOI
- Poroikov, V. (2015). 20th EuroQSAR: Understanding Chemical-Biological Interactions. Molecular Informatics, 34(6-7), 340. DOI
- Kramer, C., Kalliokoski, T., Gedeck, P. & Vulpetti, A. (2012). The experimental uncertainty of heterogeneous public K(i) data. Journal of Medicinal Chemistry, 55(11), 51655173. DOI
- Williams, A. J., Ekins, S. & Tkachenko, V. (2012). Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discovery Today, 17(13-14), 685701. DOI
- Tarasova, O. A., Urusova, A. F., Filimonov, D. A., Nicklaus, M. C., Zakharov, A. V. & Poroikov, V. V. (2015). QSAR modeling using large-scale databases: case study for HIV-1 reverse transcriptase inhibitors. Journal of Chemical Information and Modeling, 55(7), 1388-1399. DOI
- Ivanov, S. M., Lagunin, A. A. & Poroikov, V. V. (2016). In silico assessment of adverse drug reactions and associated mechanisms. Drug Discovery Today, 21(1), 58-71. DOI
- Standardizer. Retrieved March 24, 2018, from https://chemaxon.com/products/chemical-structure-representation-toolkit
- Murthy, P. K., Suneetha, V., Armakovic, S., Armakovic, S. J., Suchetan, P. A., Giri, L. & Rao, R. S. (2018). Synthesis, characterization and computational study of the newly synthetized sulfonamide molecule. Journal of Molecular Structure, 1153, 212-229. DOI
- Costa, R. A., Oliveira, K. M. T., Costam E. V. & Pinheiro, M. L. B. (2017). Vibrational, structural and electronic properties investigation by DFT calculations and molecular docking studies with DNA topoisomerase II of strychnobrasiline type alkaloids: A theoretical approach for potentially bioactive molecules. Journal of Molecular Structure, 1145, 254-267. DOI
- Sultan, M. A., Almansour, A. I., Pillai, R. R., Kumar, R. S., Arumugam, N., Armakovic, S., Armakovic, S. J. & Soliman. S. M. (2017). Synthesis, theoretical studies and molecular docking of a novel chlorinated tetracyclic: (Z/E)-3-(1,8-dichloro-9,10-dihydro-9,10-ethanoanthracen-11-yl)acrylaldehyde. Journal of Molecular Structure, 1150, 358-365. DOI
- Riju, Aikkal (2016). Phytochemical analysis, carminative, enzyme inhibitor, and anticancer activities of beta-elemene. Retrieved March 24, 2018, from DOI
- PASS Online. Retrieved March 24, 2018, from http://www.way2drug.com/passonline/
- Pogodin, P. V., Lagunin, A. A., Filimonov, D. A. & Poroikov V. V. (2015). PASS targets: ligand-based multi-target computational system based on public data and naive Bayes approach. SAR and QSAR in Environmental Research, 26(10), 783-793. DOI
- PASS targets. Retrieved March 24, 2018, from http://www.way2drug.com/passtargets/
- Lagunin, A., Ivanov, S., Rudik, A., Filimonov, D. & Poroikov, V. (2013). DIGEP-Pred: web-service for in-silico prediction of drug-induced expression profiles based on structural formula. Bioinformatics, 29(16), 2062-2063. DOI
- DIGEP Pred. Retrieved March 24, 2018, from http://www.way2drug.com/ge/
- Konova, V., Lagunin, A., Pogodin, P., Kolotova, E., Shtil, A. & Poroikov V. (2015). Virtual screening of chemical compounds active against breast cancer cell lines based on cell cycle modeling, prediction of cytotoxicity and interaction with' target=s. SAR and QSAR in Environmental Research, 26(7-9), 595-604. DOI
- Lagunin, A. A., Dubovskaja, V. I., Rudik, A. V., Pogodin, P. V., Druzhilovskiy, D. S., Gloriozova, T. A., Filimonov, D. A., Sastry, G. N. & Poroikov, V. V. (2018). CLC-Pred: a freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLOS One, 13(1), e0191838. DOI
- PASS CLC Pred, Retrieved March 24, 2018, from http://www.way2drug.com/cell-line/
- Rudik, A. V., Dmitriev, A. V., Lagunin, A. A., Filimonov, D. A. & Poroikov V. V. (2014). Metabolism sites prediction based on xenobiotics structural formulae and PASS prediction algorithm. Journal of Chemical Information and Modeling, 54(2), 498-507. DOI
- SMP. Retrieved March 24, 2018, from http://www.way2drug.com/SMP/
- Rudik, A., Dmitriev, A., Lagunin, A., Filimonov, D. & Poroikov, V. (2015). SOMP: web-service for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics, 31(12), 2046-2048. DOI
- SOMP. Retrieved March 24, 2018, from http://www.way2drug.com/SOMP/
- Rudik, A. V., Dmitriev, A. V., Lagunin, A. A., Filimonov, D. A. & Poroikov, V. V. (2016). Prediction of reacting atoms for the major biotransformation reactions of organic xenobiotics. Journal of Cheminformatics, 8, 68. DOI
- RA, Reacting Atoms. Retrieved March 24, 2018, from http://www.way2drug.com/RA/
- Dmitriev, A., Rudik, A., Filimonov, D., Lagunin, A., Pogodin, P., Dubovskaja, V., Bezhentsev, V., Ivanov, S., Druzhilovsky, D., Tarasova, O. & Poroikov, V. (2017) Integral estimation of xenobioticsТ toxicity with regard to their metabolism in human organism. Pure and Applied Chemistry, 89(10), 1449-1458. DOI
- Rudik, A. V., Bezhentsev, V. M., Dmitriev, A. V., Druzhilovskiy, D. S., Lagunin, A. A., Filimonov, D. A. & Poroikov, V. V. (2017). MetaTox: Web Application for Predicting Structure and Toxicity of XenobioticsТ Metabolites. Journal of Chemical Information and Modeling, 57(4), 638642. DOI
- MetaTox. Retrieved March 24, 2018, from http://way2drug.com/mg/
- Ivanov, S. M., Lagunin, A. A., Rudik, A. V., Filimonov, D. A. & Poroikov, V. V. (2018). ADVER-Pred - web service for prediction of adverse effects of drugs. Journal of Chemical Information and Modeling, 58(1), 8-11. DOI
- ADVER-Pred. Retrieved March 24, 2018, from www.way2drug.com/adverpred.
- Lagunin, A., Rudik, A., Filimonov, D., Druzhilovsky, D. & Poroikov, V. (2018). ROSC-Pred: web-service for rodent organ-specific carcinogenicity prediction. Bioinformatics, 34(4), 710-712. DOI
- ROSC-Pred. Retrieved March 24, 2018, from http://www.way2drug.com/ROSC/
- KinScreen, Retrieved March 24, 2018, from http://www.way2drug.com/KinScreen/
- SAR Creator. Retrieved March 24, 2018, from http://www.way2drug.com/dr/substance.php/
- Filimonov, D. A., Zakharov, A. V., Lagunin, A. A. & Poroikov V. V. (2009). QNA based УStar TrackФ QSAR approach. SAR and QSAR in Environmental Research, 20(7-8), 679-709. DOI
- Lagunin, A., Zakharov, A., Filimonov, D. & Poroikov, V. (2011). QSAR modelling of rat acute toxicity on the basis of PASS Prediction. Molecular Informatics, 30(2-3), 241-250. DOI
- Kokurkina, G. V., Dutov, M. D., Shevelev, S. A., Popkov, S. V., Zakharov, A. V. & Poroikov V.V. (2011). Synthesis, antifungal activity and QSAR study of 2-arylhydroxynitroindoles. European Journal of Medicinal Chemistry, 46(9), 4374-4382. DOI
- Zakharov, A. V., Lagunin, A. A., Filimonov, D. A. & Poroikov, V. V. Quantitative prediction of antitarget interaction profiles for chemical compounds. Chemical Research in Toxicology, 2012, 25(11) 2378-2385. DOI
- Zakharov, A. V., Peach, M. L., Sitzmann, M. & Nicklaus, M. C. (2014). QSAR Modeling of imbalanced high-throughput screening data in PubChem. Journal of Chemical Information and Modeling, 54(3), 705-712. DOI
- Fedorova, E. V., Buryakina, A. V., Zakharov, A. V., Filimonov, D. A., Lagunin, A. A. & Poroikov V. V. (2014). Design, synthesis and pharmacological evaluation of novel vanadium-containing complexes as antidiabetic agents. PLOS One, 9(7), e100386. DOI
- Hadjikakou, S. K., Ozturka, I. I., Banti, C. N., Kourkoumelis, N. & Hadjiliadis, N. (2015). Recent advances on antimony (III/V) compounds with potential activity against tumor cells. Journal of Inorganic Biochemistry, 153, 293-305. DOI
- Ajeet, Verma, M., Rani, S. & Kumar, A. (2016). Antitarget interaction, acute toxicity and protein binding studies of quinazolinedione sulphonamides as GABA1 antagonists. Indian Journal of Pharmaceutical Sciences, 78(1), 4853.
- Unnissa, S. H. & Rajan, D. (2016). Drug design, development and biological screening of pyridazine derivatives. Journal of Chemical and Pharmaceutical Research, 8(8), 999-1004. Retrieved March 24, 2018, from http://www.jocpr.com/articles/drug-design-development-and-biological-screening-of-pyridazine-derivatives.pdf
- Mansouri, K., Abdelaziz, A., Rybacka, A., Roncaglioni, A., Tropsha, A., Varnek, A., Zakharov, A., Worth, A., Richard, A. M., Grulke, C. M., Trisciuzzi, D., Fourches, D., Horvath, D., Benfenati, E., Muratov, E., Wedebye, E. B., Grisoni, F., Mangiatordi, G. F., Incisivom G. M., Hong, H., Ng, H. M., Tetko, I. V., Balabin, I., Kancherla, J., Shen, J., Burton, J., Nicklaus, M., Cassotti, M., Nikolov, N. G., Nicolotti, O., Andersson, P. L., Zang, Q., Politi, R., Beger, R. D., Todeschini, R., Huang, R., Farag, S., Rosenberg, S. A., Slavov, S., Hu, X. & Judson R. S. (2016). CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environmental Health Perspectives, 124(7), 1023-1033. DOI
- Ozturk, I. I., Yarar, S., Banti, C. N., Kourkoumelis, N., Chrysouli, M. P., Manoli, M., Tasiopoulos, A. J. & Hadjikakou, S. K. (2017). QSAR studies on antimony (III) halide complexes with N-substituted thiourea derivatives. Polyhedron, 123, 152-161. DOI