К 40-летию Института физиологически активных веществ РАН
|
Митохондрии как важная мишень при поиске новых препаратов для лечения болезни Альцгеймера и старческих деменций
Институт физиологически активных веществ Российской академии наук
142432 Черноголовка Московской обл., Северный проезд, 1;
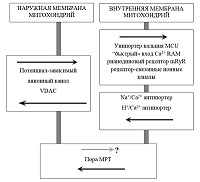
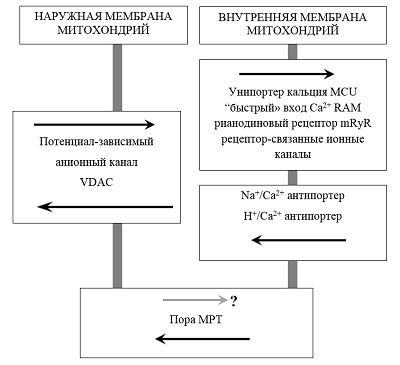
*e-mail: shevtsova@ipac.ac.ru Ключевые слова: нейродегенеративные заболевания; митохондрии; кальций; пора митохондриальной проницаемости; димебон DOI: 10.18097/BMCRM00058 ВВЕДЕНИЕ Старение является основным фактором риска спорадических форм различных нейродегенеративных заболеваний, в том числе и болезни Альцгеймера (БА). С возрастом вероятность развития БА значительно увеличивается, в 65 - 69 лет около 2% людей страдают БА, а после 90 лет – уже 25% [1]. Поэтому очевидно, что механизмы нарушения когнитивных функций при нормальном старении и при развитии нейродегенеративных заболеваний могут иметь общие черты. Одной из наиболее аргументированных и принятых в настоящее время является свободнорадикальная митохондриальная теория старения. Именно с ней тесно связаны и митохондриальные гипотезы развития спорадических форм нейродегенеративных заболеваний и, в частности, БА. Митохондриальная теория старения основывается на идее замкнутого цикла, в котором накопление с возрастом мутаций мтДНК ведёт к дисфункции дыхательной цепи, усиливая выработку радикалов кислорода, которые приводят к дальнейшему накоплению мутаций мтДНК. Возникающий биоэнергетический кризис ведет к явной дисфункции органов и дегенерации. Митохондриальная дисфункция является и одним из самых первых признаков БА [2]. Согласно митохондриальной теории БА [3] накапливающиеся с возрастом нарушения функций митохондрий, рост концентрации свободных радикалов и окислительные повреждения мембран, белков и ДНК могут вызывать чрезмерное образование Aβ и появление гиперфосфорилированной формы тау белка, провоцируя таким образом развитие БА. Важно, что внутримитохондриальное накопление амилоидных форм предшествует их внеклеточному накоплению [4]. Образующиеся в нейроне Aβ олигомеры вызывают еще большее повреждение митохондрий, увеличение окислительного стресса и провоцируют запуск каскада клеточной гибели [5]. Нарушение митохондриальных функций приводит к снижению их способности регулировать гомеостаз кальция в клетке и к снижению порога для индукции процесса скачка митохондриальной проницаемости (mitochondrial permeability transition - МРТ), который обусловлен формированием особых пор неспецифической проводимости и является ключевым этапом каскадов гибели клеток. Именно поэтому митохондрии, и процесс МРТ являются крайне перспективной мишенью для поиска нейропротекторных препаратов. Возможными механизмами увеличения устойчивости митохондрий к индукции МРТ являются ингибирование входа кальция в митохондрии, предотвращение формирования пор МРТ и/или увеличение порога их открытия, то есть увеличение кальциевой ёмкости митохондрий, причем последнее наиболее значительно в мозге. 1. ИОНЫ CA2+, ПОРЫ МРТ И ИХ РОЛЬ В НЕЙРОДЕГЕНЕРАТИВНОЙ ПАТОЛОГИИ Нарушения кальциевого гомеостаза в нейронах мозга не только закономерно происходят при старении, но и являются существенным признаком БА. Эндогенная кальций-буферная емкость интактных нейронов изменяется с возрастом, особенно в нейронах гиппокампа [6] и, в частности, в пирамидальных нейронах CA1 [7]. В значительной степени это связано с изменениями накопления и выхода кальция из внутриклеточных депо – эндоплазматического ретикулума и митохондрий [8]. Согласно литературным данным, концентрация кальция в состоянии покоя в кортикальных нейронах трансгенных мышей 3xFAD более чем в 2 раза выше, чем в нейронах мышей дикого типа (247 нмоль/л против 110 нмоль/л) [9]. Увеличение уровня кальция в клетке может провоцировать дегенеративные изменения, приводить к значительно большей вероятности запуска процесса скачка митохондриальной проницаемости с последующим запуском каскадов апоптоза и некроза [10]. Также повышенный уровень кальция способен приводить к росту генерации свободных радикалов кислорода в клетке и окислительному стрессу [11]. Важную роль в поддержании и регуляции гомеостаза Ca2+ в нейронах играют митохондрии. Митохондриальные пути транспорта кальция участвуют в регуляции таких важных функций нервных клеток, как высвобождение нейротрансмиттеров в синаптическое пространство, передача сигнала, регуляция динамики цитоскелета, адаптация биоэнергетического метаболизма к функциональной активности нейрона. Существуют многочисленные регуляторные системы поддержания гомеостаза Ca2+ в митохондриях, которые могут служить привлекательными терапевтическими мишенями для лечения БА. Транспорт Са2+ через внешнюю митохондриальную мембрану происходит в основном по потенциал-зависимому анионному каналу (VDAC). Показано, что сверхэкспрессия VDAC способствует росту входа Ca2+ в митохондрии [12]. Вход кальция в матрикс в наибольшей степени реализуется через низкоаффинный митохондриальный кальциевый унипортер (MCU), расположенный во внутренней митохондриальной мембране, кроме того, есть модель или тип «быстрого» входа кальция (RAM), митохондриальный рианодиновый рецептор (mRyR) и в последние годы обнаружен ряд рецептор-связанных ионных каналов, подобных каналам плазматической мембраны. Выход кальция из митохондрий обеспечивают Na+/Ca2+ и H+/Ca2+ антипортеры (рис. 1).
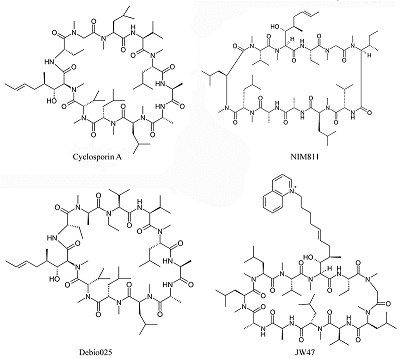
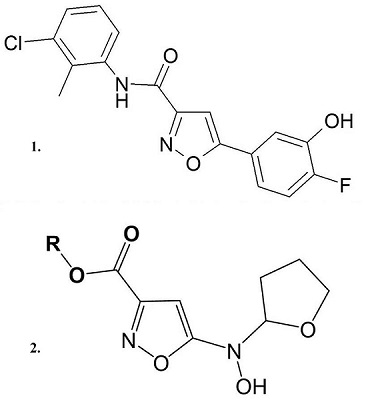
Митохондрии могут выступать в качестве эндогенных буферов вблизи плазматической мембраны или ЭР, увеличивать/уменьшать токи кальция и модулировать частоту осцилляций Са2+ в разных типах клеток [13]. В нейронах захват кальция митохондриями происходит в пресинаптических окончаниях в моменты максимальной кальциевой нагрузки, позже митохондрии медленно выбрасывают кальций и происходит его попадание в ЭР. ЭР и митохондрии тесно взаимосвязаны, поэтому изменение кальциевых функций одной органеллы приводит к нарушению передачи кальциевого сигнала при участии другой. Упомянутые ранее ассоциированные с митохондриями мембраны ЭР выполняют функции посредников в переносе фоcфолипидов, амилоида, Ca2+ и, вероятно, и других сигнальных молекул между внутриклеточными органеллами [14]. Предполагается, что MCU митохондрий взаимодействует с рецептором инозитолтрифосфата на мембране ЭР, в результате чего образуется Са2+-туннель между ЭР и митохондрией. Перенос кальция при этом происходит непосредственно из одного внутриклеточного компартмента в другой без колебаний концентрации кальция в цитозоле. В последние годы получен ряд данных о существовании в мембране митохондрий кальциевых каналов, подобных рецептор-связанным ионным каналам плазматической мембраны. Прежде всего, показано, что регуляторная ND2 субъединица NMDA рецептора кодируется митоДНК и одновременно является субъединицей комплекса I дыхательной цепи [15]. Косвенные подтверждения наличия структур, подобных NMDA-рецептору, были получены в экспериментах с влиянием лигандов этих рецепторов на митохондриальные процессы, связанные с кальциевым гомеостазом. Специфические лиганды NMDA рецептора МК801 и мемантин подавляют кальций-индуцированное МРТ и увеличивают кальциевую ёмкость митохондрий [16]. Позже было показано, что митохондрии экспрессируют белки комплекса кальциевого канала NMDA рецептора и формируют рецепторно-канальный комплекс, функционально ему идентичный. Направленная экспрессия NR1-NR2a субъединиц NMDA-рецепторного комплекса увеличивает внутримитохондриальный кальций и защищает нейрональные клетки от глутаматной токсичности [17]. Эти данные могут быть теоретическим обоснованием перспективности и реализуемости направления поиска мультитаргетных лекарственных препаратов, механизм действия которых связан одновременно с воздействием на митохондриальные кальций-зависимые функции и глутаматные рецепторы нейронов и обеспечивает улучшение когнитивных функций и нейропротекторный эффект, соответственно. Особую роль как в регуляции кальциевого гомеостаза, так и в ключевом определении жизнеспособности клетки играет процесс формирования пор МРТ, тесно связанный с процессами накопления катионов кальция митохондриями, окислительным стрессом, недостатком адениновых нуклеотидов, ростом концентрации фосфата. МРТ поры могут выступать в качестве митохондриального канала выхода Са2+ как в состоянии высокой проводимости, что в большинстве случаев сопряжено с запуском каскадов клеточной гибели, так и в состоянии низкой проводимости, способствуя функциональным изменениям кальциевого гомеостаза. Физиологическая роль этого процесса не ограничивается апоптотической элиминацией клеток в процессе развития, но также связана и с функционированием поры как быстрого канала выхода кальция из митохондрий [18]. Периодическое временное открытие-закрытие поры позволяет через изменение трансмембранного потенциала регулировать синтез ATP. Существуют гипотезы о значении поры MPT в транспорте белков по митохондриальным конгломератам и в экспрессии некоторых митохондриальных генов (например, гена белка bcl-2). Есть экспериментальные доказательства того, что механизм пресинаптической посттетанической потенциации основан на MPT и соответствующем выходе кальция из митохондриального депо. Возможно участие поры MPT в реализации синаптической пластичности и механизмах формирования памяти [19]. Активно изучается в настоящее время модуляция поры MPT различными проникающими в клетку гормонами и вторичными мессенджерами. Тем не менее, основная и наиболее изученная в настоящее время роль поры MPT – это элиминация повреждённых митохондрий, а затем и клеток, которые становятся нежелательными для нормального функционирования организма, в частности, если в них накапливаются свободные радикалы [20]. Именно митохондриям принадлежит решающая роль в запуске как каспаз-зависимого, так и каспаз-независимых путей клеточной гибели. Вопрос молекулярной структуры поры МРТ и механизмов ее регуляции, имеющий важное значение при определении молекулярных мишеней для скрининга модуляторов поры, в настоящее время остается открытым. В современной научной литературе можно встретить несколько гипотез структуры поры МРТ. Большинство из них сходятся в том, что пора имеет белковую природу. Однако ряд авторов предполагает, что пора МРТ может также иметь небелковую природу. Например, есть гипотеза, что хотя бы частично, пора МРТ образована комплексом, состоящим из поли-R-3-гидроксибутирата, полифосфатов и катионов кальция (PHB/polyp/Ca2+ комплекс). Данный комплекс, выделенный из митохондрий печени крыс, обладает свойствами, схожими со свойствами поры МРТ [21]. Важно отметить, что полифосфат играет не последнюю роль в митохондриальном метаболизме и в процессах накопления Ca2+ митохондриями. Экспериментально показано, что снижение уровня полифосфата увеличивает кальциевую ёмкость митохондрий и снижает вероятность вызванного Са2+ открытия поры МРТ [22]. В рамках гипотезы о строении поры как мультибелкового образования также не существует ясности на настоящий момент о конкретных компонентах поры. Выделяют три основных предполагаемых компонента поры МРТ: потенциал-зависимый анионный канал (VDAC), переносчик адениновых нуклеотидов (ANT) и циклофилин Д (CypD). Данная модель поры была подтверждена с использованием очищенных белков-компонентов в липосомах, причем, циклоспорин А (наиболее известный ингибитор МРТ, лиганд циклофилина Д) ингибировал образование поры МРТ-подобных пор в липосомах. Изначально предполагалось, что основными порообразующими компонентами являются белки VDAC и ANT. Однако ряд экспериментальных данных позволяет заключить, что оба белка не являются её абсолютно необходимыми ключевыми составляющими. На трансгенных мышах, полных нокаутах по трём изоформам VDAC [23] или нокаутах по двум изоформам ANT было показано, что даже в отсутствии любых изоформ этих белков митохондрии обладают порообразующей активностью и чувствительностью к циклоспорину А. В то же время, экспериментальные доказательства позволяют предположить, что в образовании и регуляции поры играют роль и другие белки, в том числе проапоптотические Bax белки, антиапоптотические белки семейства Bcl, гексокиназа, митохондриальный фосфатный переносчик [24], периферический бензодиазепиновый рецептор [25] и комплекс I дыхательной цепи митохондрий [26]. Сравнительно недавно было показано, что локализованная во внутренней митохондриальной мембране ATP-синтаза (комплекс V ДЦ митохондий) тоже может являться потенциальным компонентом поры МРТ. Известно, что ATP-синтаза способна димеризоваться. Димеры, вероятно, вызывают формирование крист митохондрий и оптимизируют каталитическую функцию фермента, но, кроме того, именно димеры ATP-синтазы, но не ее мономеры, могут проявлять порообразующую активность, регулируемую Ca2+, Mg2+/ADP, Pi и другими модуляторами МРТ [27]. Циклофилин Д связывается с ATP-синтазой в присутствии неорганического фосфата, при этом происходит частичное ингибирование ATP-синтазной функции фермента и уменьшается порог чувствительности митохондрий к открытию поры. Образование комплекса циклоспорина А (ЦсА) с циклофилином Д препятствует связыванию последнего с ATP-синтазой и, соответственно, увеличивает её ферментативную активность и снижает порообразующую активность, увеличивая порог чувствительности митохондрий к индукции МРТ. В литературе также есть данные о том, что ATP-синтаза взаимодействует и с другими ключевыми участниками процесса скачка митохондриальной проницаемости - с ANT и с митохондриальным переносчиком фосфата. Важную роль в процессе вызванного ростом концентрации катионов кальция ЦсА-зависимого открытия поры МРТ играет с-субъединица F0-ATP-синтазы [28]. В то же время, в клеточной линии, полностью лишённой всех изоформ этой субъединицы, наблюдается индукция процесса МРТ и кальциевая ёмкость митохондрий не отличается от контрольных клеток [29], что не позволяет считать с-субъединицу F0-ATP-синтазы абсолютно необходимым компонентом поры МРТ. Cуществует и гипотеза о том, что пора МРТ образована мембранными белками с нарушенным вследствие окислительного и прочих видов стресса фолдингом, и ее регулируют шапероноподобные белки (к которым относится и циклофилин Д) [30]. Циклофилин Д блокирует проводимость через эти белковые агрегаты, но когда число подобных белковых кластеров превышает количество циклофилина Д, происходит нерегулируемое открытие пор, стимулируемое кальцием и ингибируемое связыванием ЦсА с циклофилином Д. Таким образом, все предполагаемые в настоящее время модели состава поры МРТ имеют как очевидные доказательства своего существования, так и доказательства возможности альтернативного пути формирования поры. Это позволило нам предположить, что пора МРТ является динамичным ансамблем с переменным составом, который может варьироваться в зависимости от типа ткани, триггеров, обуславливающих ее открытие, внутриклеточных условий в данный момент времени и прочих факторов. Поскольку не существует единой теории строения поры МРТ, каждый из вероятных ее компонентов может рассматриваться как потенциальная мишень новых лекарственных препаратов. И в то же время, именно процесс МРТ, регистрируемый как кальций-индуцируемое "набухание", деполяризация митохондрий или исчезновение кальций-буферной активности митохондрий может быть перспективной методической основой для скрининговых исследований потенциальных ингибиторов МРТ. 2. ИНГИБИТОРЫ МРТ КАК ПЕРСПЕКТИВНАЯ ГРУППА ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ ЛЕЧЕНИЯ ЗАВИСИМОЙ ОТ ВОЗРАСТА НЕЙРОДЕГЕНЕРАТИВНОЙ ПАТОЛОГИИ Коррекцию митохондриальных функций и митохондриального кальциевого гомеостаза можно рассматривать в качестве перспективных подходов ранней терапии нейродегенеративных заболеваний, и одними из наиболее перспективных из этого класса препаратов могут быть ингибиторы (модуляторы) процесса МРТ (и как препараты, увеличивающие выживаемость нейронов, и как препараты способные поддержать функциональную активность синапсов благодаря увеличенной кальциевой ёмкости митохондрий). Одними из первых препаратов такого класса явились аналоги специфического и высокоэффективного (IC50=0.5нМ) ингибитора МРТ ЦсА (рис. 2), благодаря которому были проведены исследования структуры, регуляции, специфичности поры МРТ, её участие во многих патологических процессах и заболеваниях, показано, что ингибирование поры МРТ может иметь положительный терапевтический эффект при ряде дегенеративных заболеваний [31,32]. Но ЦсА обладает иммуносупрессивным действием и, соответственно, большим числом побочных действий, а кроме того, активно секвестрируется эритроцитами, плохо проникает через гематоэнцефалический барьер [33]. В связи с этим были разработаны его неиммунносупрессивные производные именно как ингибиторы МРТ: N-метил-4-изолейцин-циклоспорин (NIM811) (рис. 2), который обладает цитопротекторным действием на культуре гепатоцитов крыс, а также проявил нейропротекторное действие после травматического повреждения мозга и ишемии мозга [34,35]; Debio025 (рис. 2), который не только ингибирует индукцию МРТ и снижает интенсивность некротической гибели клеток, но также оптимизирует кальциевые сигналы в состоянии стресса у mdx мышей (модели миодистрофии Дюшенна) [36]; JW47 (рис. 2), хинолин-катион модифицированный ЦсА с увеличенным сродством к митохондриям и проявляющий нейропротекторные свойства в экспериментальной модели множественного склероза [37]. Действие ЦсА и его производных на процесс МРТ обусловлено связыванием с компонентом (или, по некоторым теориям, модулятором) МРТ циклофилином Д – белком митохондриального матрикса – и ингибированием его пептидилпролил-изомеразной активности уже в наномолярных концентрациях.
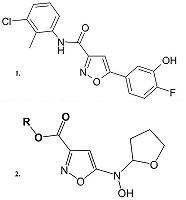
Недавно в результате программ направленного скрининга высокоэффективных ингибиторов МРТ был выявлен ряд превосходящих ЦсА ингибиторов МРТ, в частности, диарилизоксазол-3-карбоксамиды, которые повышают устойчивость митохондрий печени мышей к Са2+ индуцированному открытию поры МРТ и увеличивают более чем в 20 раз кальциевую емкость митохондрий, не оказывая влияние на митохондриальный потенциал вплоть до концентрации 100 мкМ. Действующие концентрации диарилизоксазол-3-карбоксамидов ниже, чем у ЦсА, причём, основываясь на синергичности действия этих ингибиторов, авторы делают вывод об отличной от ЦсА молекулярной мишени действия этих соединений [38]. В результате оптимизации структур авторам удалось создать ряд соединений, которые в пикомолярных концентрациях ингибируют открытие пор МРТ. Для соединения-лидера из этого ряда (N-(3-хлоро-2-метилфенил)-5-(4-фтор-3-гидроксифенил)изоксазол-3-карбоксамид) (рис. 3) в экспериментах in vivo наблюдался терапевтический эффект на данио-рерио модели одного из дегенеративных заболеваний - врожденной мышечной дистрофии Ульриха [38]. Эти соединения представляют значительный интерес для дальнейших испытаний в качестве потенциальных нейропротекторов. Для соединений, также содержащих изоксазольный и адамантановый фрагменты (рис. 3) было показано наличие радикал-связывающей активности, способности подавлять ПОЛ и ингибировать липоксигеназу, а также увеличение в их присутствии скорости поглощения кислорода изолированными митохондриями печени крыс как в сопряженном, так и в разобщенном состоянии при отсутствии влияния на митохондриальный мембранный потенциал и отсутствии цитотоксичности [39]. Эти in vitro характеристики также позволяют предположить потенциальный нейропротекторный потенциал.
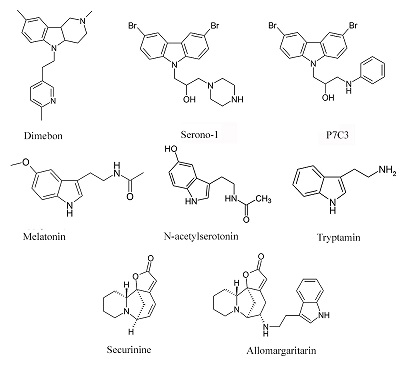
Окислительный стресс в значительном числе случаев рассматривается как условие сниженной устойчивости митохондрий к открытию пор МРТ. В соответствии с этим разработан целый ряд потенциальных лекарственных препаратов-антиоксидантов, обладающих способностью накапливаться в митохондриях [40]. Для ряда подобных препаратов показана их способность подавлять чувствительность митохондрий к индукции МРТ [41], и именно эти препараты представляются нам особенно интересными в качестве основы для создания лекарственных препаратов для лечения нейродегенеративных заболеваний. Одним из наиболее привлекательных направлений создания подобных лекарственных средств является разработка аналогов эндогенных антиоксидантов и регуляторов МРТ. Гормон эпифиза мелатонин (рис. 4), являющийся эндогенным антиоксидантом с радикал-связывающей активностью и способностью ингибировать активность NO-синтазы, обладает значительным нейропротекторным потенциалом [42,43]. Более того, этот индоламин помимо антиоксидантного и рецептор-опосредованного гормонального эффекта влияет и на ряд митохондриальных функций. Он действует как стимулятор дыхательной цепи митохондрий, увеличивая активность комплексов I и IV и специфически накапливаясь в митохондриях [44]. В последнее время появились данные о непосредственном ингибировании процесса МРТ мелатонином в экспериментах на клеточной культуре и при исследовании методом патч-кламп изолированных митохондрий мозга – IC50 = 0.8 мкМ [45]. Эти данные позволяют предположить, что наблюдаемый положительный эффект при ряде нейродегенеративных заболеваний связан не только с гормональным и антиоксидантным эффектом, регуляцией циркадных ритмов и нормализацией сна мелатонином, но и с направленным действием на митохондрии и, в частности, со способностью ингибировать процесс МРТ. Предшественник мелатонина, N-ацетилсеротонин (NAS) (рис. 4), во многом обладает сходными с мелатонином свойствами: нейропротекторными, антивозрастными свойствами [46]. Но в отличие от мелатонина синтезируется и обнаруживается в наномолярных концентрациях в ряде отделов головного мозга, обладает более выраженным антиоксидантным потенциалом и так же, как мелатонин, проявляет нейропротекторный эффект, ингибируя процесс МРТ на клеточном уровне и митохондриальный этап клеточной гибели [47]. В то же время, показано отсутствие прямого подавления МРТ в изолированных митохондриях в диапазоне концентраций NAS от 1 до 30 мкМ, впрочем, как и непосредственное влияние мелатонина на открытие пор МРТ [48]. В этом диапазоне концентраций вышеописанные результаты согласуются с полученными нами ранее. Однако нами было показано, что ингибирующее влияние NAS на МРТ в изолированных митохондриях наблюдается в более низком диапазоне концентраций 5 - 50 нМ и наиболее стабильно проявляются при индукции открытия пор МРТ нейротоксином MPP+ [49]. Такая неоднозначность действия мелатонина и в большей степени NAS, немонотонная зависимость от концентрации и условий индукции МРТ достаточно хорошо согласуется с гипотезой о том, что это соединение играет особую роль как эндогенный и локальный регулятор жизнеспособности нейронов и процесса МРТ.
Cтруктурный аналог и предшественник мелатонина триптамин (рис. 4) обладает незначительной антиоксидантной активностью (IC50 = 0,84 мМ) и не проявляет свойств ингибитора МРТ [50], однако описанный нами его конъюгат с алкалоидом секуринином (также не обладающим этими типами активностей) – алломаргаритарин (рис. 4) в микромолярном диапазоне концентраций проявляет высокую антиоксидантную активность, которая связана с его способностью хелатировать ионы Fe(II) и с антирадикальной активностью. Кроме того, алломаргаритарин эффективно блокирует открытие пор МРТ, в том числе и индуцированное Аβ-пептидом [51]. Выбор секуринина для создания потенциального лекарственного препарата основывался на имеющихся данных о его активности как стимулятора когнитивных функций, в том числе и в условиях Aβ-вызванной токсичности. Молекулярными механизмами этих эффектов может быть роль секуринина как антагониста ГАМК рецепторов и ингибирование ацетилхолинэстеразы [52,53]. Алломаргаритарин проявляет выраженные нейропротекторные свойства, защищая нейроны первичной культуры коры мозга от гибели, вызванной эксайтотоксичностью, окислительным стрессом и Аβ. Очевидно, этот эффект связан с его антиоксидантной и митопротекторной активностью [54,55]. Кроме того, алломаргаритарин снижает агрегацию Аβ [56]. Противосудорожная активность алломаргаритарина на моделях индуцированных пилокарпином судорог [57], также может быть отчасти связана с его способностью увеличивать устойчивость митохондрий к индукции МРТ. О роли МРТ в противосудорожном эффекте кетоновых тел и топирамата достаточно убедительно свидетельствуют исследования на трансгенных мышах Kcna1 со спонтанной эпилептической активностью и пилокарпиновой модели хронических судорог, соответственно [58,59]. Биоизостерным аналогом мелатонина можно считать и препарат димебон (рис. 4), который был разработан и запатентован как неселективное антигистаминное лекарственное средство учеными МГУ в начале 80-х годов прошлого века [60]. Позже было установлено, что димебон проявляет когнитивно стимулирующие и нейропротекторные свойства на нейротоксикологических и трансгенных моделях ряда нейродегенеративных заболеваний и является модулятором поры МРТ, увеличивая устойчивость к её индукции. Уникальной особенностью димебона можно назвать мультинаправленность его действия, сочетание когнитивно-стимулирующего и нейропротекторного эффектов с соответствующим сочетанием мишеней. Когнитивно-стимулирующий эффект препарата связывают, в основном, с ингибированием ряда нейрональных рецепторов и ионных каналов, в том числе NMDA-рецепторов, серотониновых 5-HT7 рецепторов и потенциал-зависимых кальциевых каналов L-типа. Нейропротекторный потенциал димебона может во многом основываться на его способности увеличивать функциональную устойчивость митохондрий. На изолированных митохондриях печени и мозга крыс было показано, что димебон подавляет индукцию МРТ различными стимулами (ионами фосфата, кальция или трет-бутилгидроксипероксидом – тБГП), и, в отличие от ЦсА, проявляет свойства антиоксиданта, снижая перекисное окисление липидов митохондрий печени крыс, вызванное тБГП или Aβ [61]. В то же время димебон является гораздо более слабым модулятором МРТ поры, чем ЦСА, и проявляет свой эффект только в состоянии энергизации митохондрий, то есть в условиях работы дыхательной цепи митохондрий. Ингибирование индукции МРТ димебоном сопровождается увеличением кальциевой емкости митохондрий [62], что вносит свой вклад в когнитивно-стимулирующие свойства димебона, нормализуя синаптическую нейротрансмиссию. Нейропротекторный эффект димебона широко подтверждается на клеточных культурах: как защита от глутаматной эксайтотоксичности, Aβ-, МРР+-, AF64A-, тБГП- и иономицин-вызванной токсичности и т.п. С использованием первичной культуры нейронов коры головного мозга мышей и клеток нейробластомы человека SH-SY5Y было показано, что наномолярные концентрации димебона увеличивают активность сукцинатдегидрогеназы, стабилизируют митохондриальный трансмембранный потенциал (∆Ψm) и увеличивают уровень АТР в клетке [63]. При этом димебон не влияет на количество митохондриальной ДНК, то есть не индуцирует биогенез митохондрий. В этой же работе было показано, что в условиях стресса предобработанные димебоном нейроны более устойчивы к повышенной внутриклеточной концентрации кальция, в меньшей степени подвержены деполяризации митохондрий в отличие от контрольных нейронов. Димебон в наномолярных концентрациях ингибирует апоптоз клеток нейробластомы человека SH-SY5Y [64] и нивелирует негативное действие сверхэкспрессии мутантной формы Aβ (HEKsw) на морфологию митохондрий и активность ферментативных комплексов дыхательной цепи митохондрий [65]. Кроме того, димебон способен увеличивать сниженную в этих клетках экспрессию белка TIMM (транслоказы внутренней мембраны митохондрий), который обеспечивает транспорт ядерно-кодируемых белков, в том числе и участвующих в формировании комплексов дыхательной цепи, через внутреннюю мембрану митохондрий [66]. Учитывая данные о митопротекторном и антиоксидантном эффектах димебона мы предположили и затем экспериментально подтвердили в опытах in vivo геропротекторные свойства димебона [67]. Димебон оказывает положительное влияние на метаболизм мозга стареющих крыс, в том числе и нормализуя потребление глюкозы [68]. Несмотря на имеющиеся в литературе данные об отсутствии снижения нерастворимых амилоидных агрегатов в мозге трансгенных животных с 5xFAD (модель БА), даже в этом исследовании была выявлена коррекция поведенческих нарушений – снижение тревожности у этих животных под влиянием димебона [69]. Кроме того, для ряда трансгенных моделей протеинопатий как моделей нейродегенеративных заболеваний показано, что хроническое введение димебона или его фтор-содержащих аналогов вызывает снижение специфической патологии при одновременном снижении числа нерастворимых агрегатов связанных с этими нейродегенеративными заболеваниями белков [70,71]. Снижение агрегатов наблюдается и на клеточных моделях протеинопатий [72] и, вероятно, в значительной степени связано с усилением аутофагии [73]. Подобное сочетание активностей – ингибирование МРТ, активация аутофагии и соответствующее снижение агрегатов – наблюдали для 4-азастероидов и, возможно, именно умеренное снижение числа митохондрий с активированной порой МРТ в присутствии азастероидов и димебона приводит к снижению вероятности активации апоптоза или некроза в клетках и превалированию активации локальной аутофагии. Структурно близкими для мелатонина и димебона соединениями можно считать и аминопропилкарбазольные производные, описанные в работе [74] и проявляющие пронейрогенную активность, защищая новые (newborn) нейроны от апоптоза благодаря митопротекторной активности. Стимулируют нейрогенез в гипокампе также димебон и бром-содержащий карбазол Serono-1, причем авторы отмечают корреляцию между митопротекторным эффектом, нейропротекцией и пронейрогенной активностью: наиболее активное соединение во всех тестах – соединение-лидер P7C3 (рис. 4) в ряду аминопропилкарбазолов, менее активны Serono-1 (рис. 4) и димебон. Как в вопросах выживаемости-гибели клеток, так и в вопросе нейрогенеза, то есть дифференцировки и дальнейшего выживания вновь образованных нейронов, митохондрии играют крайне важную роль. Дифференцировка полипотентных клеток в нейроны сопровождается переключением биоэнергетики клетки с анаэробного гликолиза на аэробное окислительное фосфорилирование [75]; и образующиеся при работе дыхательной цепи в результате кратковременного локального открытия пор МРТ «вспышки» (flash) образования супероксид-анионов, по-видимому, являются одним из механизмов регуляции дифференцировки этих клеток. В зависимости от частоты flash-открытия МРТ и выброса супероксид-аниона клетка либо включает каскад дифференцировки, либо реагирует на повышение свободных радикалов супероксид-анионов запуском апоптоза или некроза [76]. Но, тем не менее, ингибиторы МРТ, и вышеперечисленные, и ЦсА, стимулируют нейрогенез в большей степени благодаря увеличению выживаемости вновь дифференцированных клеток [77]. Ингибирование МРТ и, возможно, активация никотинамид фосфорибозилтрансферазы - скорость-лимитирующего фермента каскада образования никотинамид адениновых динуклеотидов [78], а также стимулирование нейрогенеза обуславливают когнитивно-стимулирующий и нейропротекторный потенциал этой группы соединений в целом ряде неврологических и нейродегенеративных заболеваний: ишемический инсульт, травматическое повреждение мозга, болезнь Паркинсона и Альцгеймера, боковой амиотрофический склероз [79,80]. ЗАКЛЮЧЕНИЕ Практически все описанные в данном обзоре соединения, являясь ингибиторами/модуляторами поры МРТ и в соответствии с этим проявляющие нейропротекторный эффект, взаимодействуют с другими мишенями, обеспечивающими сопряженные положительные терапевтические эффекты – стимулирование когнитивных функций, противосудорожный, антидепрессатный и т.д. То есть фактически все описанные соединения являются мультитаргетными препаратами. Разработка новых высокоэффективных препаратов для лечения широкого спектра нейродегенеративных заболеваний является одним из самых сложных и востребованных направлений современной медицинской химии. Поиск и дизайн многоцелевых препаратов, действующих одновременно на несколько мишеней, участвующих в патогенезе этих заболеваний, в настоящее время считается одной из наиболее перспективных стратегий [81,82]. В связи с этим использование митохондрий как основы для формирования скрининговой стратегии поиска соединений для лечения нейродегенеративных заболеваний представляет особый интерес – и как тестирование их потенциальной токсичности, и как основа для создания метаболических стимуляторов и препаратов, обладающих нейропротекторным и когнитивно-стимулирующим действием. БЛАГОДАРНОСТИ Работа выполнена в рамках Госзадания 0090-2017-0019. ЛИТЕРАТУРА
|