|
СОДЕРЖАНИЕ |
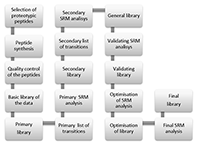
Алгоритм создания таргетных масс-спектрометрических методов для детекции белков Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121 Москва, ул. Погодинская, 10 стр. 8, *e-mail: victor.zgoda@gmail.com Ключевые слова: мониторинг выбранных реакций; пептиды; стабильные изотопно-меченные стандарты; масс-спектрометрии DOI:10.18097/BMCRM00006 ВВЕДЕНИЕ В настоящее время самым информативным инструментом для анализа белков в биологических объектах является протеомика с использованием масс-спектрометрии [1] . Если ранее основной целью масс-спектрометрии в протеомных исследованиях являлась идентификация белков, то в настоящее время приоритетными становятся именно количественные измерения. В клинической лабораторной диагностике определение концентраций различных белков в биологических жидкостях имеет особенно важное значение [2]. Традиционно для определения концентраций белков применяются иммуноферментные методы, такие как ELISA. Однако, развивающиеся в настоящее время методы масс-спектрометрии и, особенно, методы направленного анализа (SRM) являются конкурирующими подходами проведения анализов по определению содержания белков [3]. Измерения абсолютных концентраций белков в масс-спектрометрии стали возможны благодаря внедрению в практику внутренних стандартов, которые в точности соответствуют химической структуре измеряемого вещества, но синтезированы с использованием изотопно-меченых молекул. Такие изотопно-меченые внутренние стандарты (Isotopically labeled internal standard, SIS), добавляемые к аналитам в известных концентрациях, позволяют учитывать все возможные ошибки измерения концентраций, начиная от девиаций в количестве наносимой пробы и заканчивая нестабильностью или особенностями ионизации анализируемого соединения. Особенностью данного подхода является его высокая специфичность, которая обеспечивается измерением не только массы пептида, специфичного для определенного белка, но и его фрагментов, подтверждающих искомую аминокислотную последовательность. Считается, что в сочетании с использованием изотопно-меченых внутренних стандартов SRM превосходит иммуноферментные методы по специфичности и точности количественной оценки белков [2]. Данный подход является направленным и подразумевает выбор специфичного для каждого белка стандарта и создание соответствующей ему методики [4]. Из вышесказанного понятно, что от того, насколько правильно будет выбран внутренний стандарт, зависит качество идентификации и измерения концентрации таргетного белка. В данной работе суммирован наш опыт по выбору пептидного стандарта для измерения концентрации белков. Результаты представлены в виде алгоритма, который учитывает основные стадии выбора внутреннего стандарта и основные моменты разработки методов детекции и определения концентрации белков. МАТЕРИАЛЫ И МЕТОДЫ Стандарты пептидов получали твердофазным пептидным синтезом на приборе Overture (Protein Technologies, США) согласно методике, опубликованной в работе Hood с соавт. [5]. При синтезе изотопно-меченых пептидов вместо обычного лейцина (Fmoc-Leu-OH) использовали изотопно-меченый (Fmoc-Leu-OH-13C6,15N или Fmoc-Lys-OH-13C6,15N2). Количественное определение проводили методом внутреннего стандарта, в качестве которого использовали изотопно-меченые аналоги нативных пептидов. В работе использовали пептиды, приведенные в приложении. При создании опорной библиотеки разделение пептидов осуществляли на хроматографической системе Ultimate 3000 RSLCnano («Thermo Scientific», США). Пептиды разделяли на колонке Acclaim Pepmap® C18 (75 мкм х 150 мм, 2 мкм размер частиц) («Thermo Scientific») в градиенте подвижной фазы А (0,02% уксусная кислота, 0,1% муравьиная кислота, рН 2,66) и подвижной фазы Б (80% ацетонитрил, 0,02% уксусная кислота, 0,1% муравьиная кислота) при скорости потока 0,3 мкл/мин. Перед аналитическим разделением пептиды наносили на обогащающую колонку Accalaim µ-Precolumn (0,5 мм х 3 мм, 5 мкм размер частиц) («Thermo Scientific») при скорости потока 10 мкл/мин в течение 5 минут в изократическом режиме подвижной фазы В (2% ацетонитрил, 0,02% уксусная кислота, 0,1% муравьиная кислота, рН 2,63). Разделение проводили в линейном градиенте от 2% до 37% подвижной фазы Б в течение 55 мин, концентрацию буфера Б линейно повышали до 90% за 5 мин, далее промывали колонку при 90% подвижной фазы Б в течение 10 мин, снижали содержание буфера Б до 2% за 5 мин и уравновешивали в начальных условиях градиента при 2% подвижной фазы Б в течение 15 мин. Масс-спектрометрический анализ проводили на гибридном орбитальном масс-спектрометре Orbitrap Fusion («Thermo Scientific») в режиме положительной ионизации в источнике NSI («Thermo Scientific»). Температура осушающего газа (азот) составляла 280оС, напряжение на эмиттере 2.1 кВ. Панорамное сканирование проводили в диапазоне масс от 400 m/z до 1200 m/z, тандемное сканирование фрагментных ионов - от нижней границы 110 m/z до верхней границы, определяемой зарядным состоянием прекурсорного иона, но не более 2100 m/z. Для тандемного сканирования учитывали только ионы от z = 2+ до z = 6+ по зарядному состоянию. Максимальное число разрешённых для синхронной изоляции ионов в режиме MS2 было установлено не более 20. Максимальное время накопления для прекурсорных ионов составило не более 50 мс для фрагментных ионов не более 110 мс. Cоздание и валидацию SRM метода осуществляли на масс-спектрометрическом детекторе “Agilent 6490” QQQ в режиме SRM c источником ионизации “JetStream” при температуре осушающего газа 250оС (скорость потока 14л/мин), фокусирующего газа - 280оС (скорость потока 11 л/мин) и давлении на распылителе 1,15 атм. Напряжение на входном капилляре - 3,5 кВ; фокусирующее напряжение 450 В. Хроматографическое разделение проводили при помощи высокоэффективного жидкостного хроматографа Infinity 1290 (“Agilent”, США), как описано ранее [6] в градиенте подвижной фазы А (водный раствор 0,1% муравьиной кислоты и 0,01% трифторуксусной кислоты) и подвижной фазы Б (ацетонитрил с 0,1% муравьиной кислотой и 0,01% трифторуксусной кислотой) на хроматографической колонке Zorbax Eclipse C18 (2,1 х 50 мм, 1,8 мкм размер частиц, 100А размер пор). Содержание подвижной фазы Б возрастало с 3% до 8,5% в течение 2-х минут, затем линейно возрастало до 32% в течение последующих 48-ми минут, после чего содержание фазы Б увеличивали до 53.6% в течение последующих 2-х минут и до 80% в течение последующей одной минуты. Колонку промывали в изократическом режиме 80% фазой Б в течение 5-ти минут, после чего систему возвращали к начальным условиям градиента элюции (3% Б) в течение 2-х минут. Уравновешивание системы проводили в начальных условиях (3% В) в течение 6-ти минут. Скорость потока сохраняли постоянной на уровне 0,3 мл/мин с постоянным термостатированием колонки при температуре 42оС. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Разработка метода SRM для регистрации и количественной оценки белков по протеотипическим пептидам проходит несколько стадий, обозначенных на рисунке 1. Каждый из этапов имеет внутренние критерии отбора пептидов, их фильтрации, верификации и последующего составления метода количественной оценки белков с использованием масс-спектрометра типа тройного квадруполя. Выбор пептидов из списка протеотипических пептидов, синтез пептидов Первичный этап разработки включает в себя составление списка протеотипических пептидов. Регистрация конкретного пептида в протеомном эксперименте зависит от многих факторов, связанных с физико-химическими свойствами самого пептида, а также от биологической матрицы, в которой проводятся измерения. Поэтому, на первом этапе выбора пептидов логичным является использование экспериментальных данных. Полезными ресурсами для выявления ранее зарегистрированных пептидов белков являются открытые протеомные ресурсы и базы данных, такие как: Human Protein Atlas, PRIDE, PeptideAtlas и SRMatlas. Если искомый белок не был ранее зарегистрирован и отсутствует в списках вышеупомянутых протеомных ресурсов, для составления списка можно воспользоваться одним из многочисленных сервисов по предсказанию протеолических пептидов, исходя из известной структуры белка и специфичности используемой протеазы (напр. см. https://web.expasy.org/peptide_mass/, Skyline). Основным критерием выбора является уникальность аминокислотной последовательности пептида для исследуемого белка в пределах биологического вида, на котором проводятся исследования. В зависимости от поставленной цели, пептид для SRM может быть специфичным либо для канонической формы белка, либо для одной из его изоформ, если ставится задача проводить изоформа-специфичный протеомный анализ. Кроме того, согласно требованиям HUPO, специфичными считаются пептиды имеющие 9 и более аминокислотных остатков. Удачным ресурсом для определения специфичных последовательностей пептидов является программное обеспечение Peptide unicity checker (https://www.nextprot.org/viewers/unicity-checker/app/index.html). Отобранные уникальные пептиды проходят последующий отбор для составления первичного списка для химического синтеза. В основе отбора пептидов для синтеза лежат несколько критериев, одним из которых является обязательное присутствие в составе аминокислотной последовательности остатков, для которых имеются коммерчески доступные производные аминокислот с включенными стабильными изотопами углерода и азота (например, лейцин, серин, глицин, аргинин или лизин). Если планируется использование трипсина в качестве гидролитического фермента, то лизин или аргинин могут быть только C-терминальными аминокислотными остатками, так как пептиды, отбираемые для химического синтеза и разработки SRM метода, должны отвечать требованиям специфичности ферментативной реакции, проводимой на этапе подготовки биологической пробы. Выбор пептидов, в составе которых в обязательном порядке должны присутствовать вышеуказанные аминокислотные остатки, обусловлен тем, что при химическом синтезе именно эти аминокислоты в своём составе будут иметь стабильные тяжёлые изотопы углерода 13С и азота 15N.
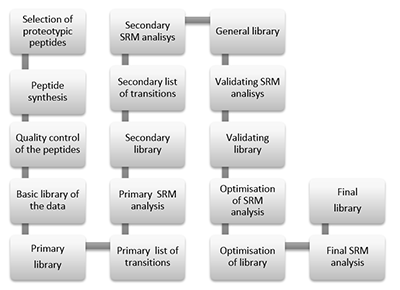
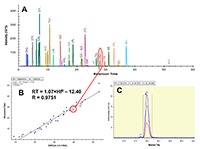
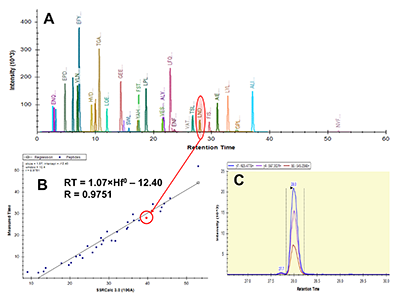
Пептиды, в составе которых будут присутствовать аминокислоты, содержащие тяжёлые изотопы углерода и азота, выступают в качестве внутренних стандартов. Они идентичны по своим физико-химическим свойствам (включая хроматографическую подвижность) эндогенным пептидам с аналогичной аминокислотной последовательностью, но отличаются от них по изотопному составу. Однако, на первичном этапе химического синтеза отобранных пептидов предпочтительно синтезировать т.н. «легкие» пептиды, идентичные эндогенным по изотопному составу. Дополнительными критериями отбора пептидов для химического синтеза из общего списка протеотипических пептидов являются длина пептида (не менее 9 а.о., но не более 25 а.о.), теоретическая гидрофобность пептида (в пределах от 10 до 60 относительных единиц), а также желательное отсутствие в составе пептида аминокислотных остатков метионина и цистеина. Пределы длины пептида обусловлены, главным образом, требованиям химического синтеза, минимизацией вырожденности последовательности и требованиями масс-спектрометрической детекции (пептиды малой длины будут регистрироваться в динамической области, насыщенной и характерной для низкомолекулярных органических соединений непептидной природы). В конечном итоге из общего списка пептидов с учётом указанных критериев для каждого исследуемого белка следует отобрать не более 3-х пептидов. Проверка качества (подлинности) синтетических пептидов. Составление опорной спектральной библиотеки. Химический синтез отобранных пептидов проводится для определения их масс-спектрометрических характеристик с последующим выбором основного пептида для его синтеза с включением стабильных изотопов углерода и азота. Данный пептид будет выступать в роли стандарта измерения в окончательных методах SRM. На этапе первичного синтеза пептиды могут быть объединены в смеси по 50 - 96 штук. Пептиды не очищены от побочных продуктов химического синтеза, парциальная концентрация каждого пептида и суммарная концентрация в пептидной смеси не известны; эти данные не играют решающей роли в составлении опорной библиотеки спектров для выбора стандартного пептида. Для составления опорной спектральной библиотеки смесь пептидов предварительно анализируют на масс-спектрометре высокого (типа Q-ToF) или сверхвысокого (типа Orbitrap) разрешения в ходе хроматографического разделения пептидной смеси. В результате на выходе получаем 1-2 файла данных для анализа(рис. 2).
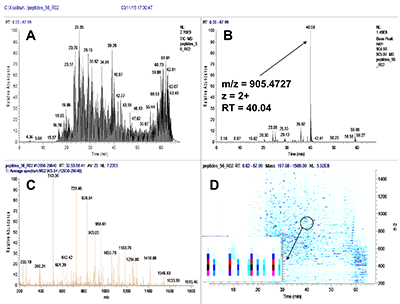
Для составления опорной библиотеки спектров можно использовать программное обеспечение Skyline, в котором проводится первичная обработка полученных результатов. Процесс обработки данных и составления опорной библиотеки начинается с загрузки в программу Skyline файла (в формате fasta, см. файл 56X13.fasta в дополнительных материалах) с информацией о полной аминокислотной последовательности белков, пептиды которых были синтезированы и проанализированы на масс-спектрометре высокого разрешения. Параметры в программном обеспечении устанавливаем таким образом, чтобы информация о загруженных белках была представлена в виде набора всех возможных пептидов, которые могут быть продуктами ферментативной реакции расщепления с участием трипсина. Наряду с информацией о длине и специфичности, также устанавливается информация о пределах длины пептидов (от 9 до 25 а.о.) и информация о возможных зарядных состояниях пептидных ионов (в пределах от z = 2+, 3+). При загрузке файла масс-спектрометрических данных, во-первых, проводится автоматизированная детекция сигнала. Сигналы, которым не был присвоен какой-либо из предполагаемых в смеси пептидов, исключаются из дальнейшего анализа, таким образом, остаются пептиды, для которых был присвоен атрибут найденного в спектральной библиотеке сигнала детекции (рис. 3, см. X13_56_light_basic_lib).
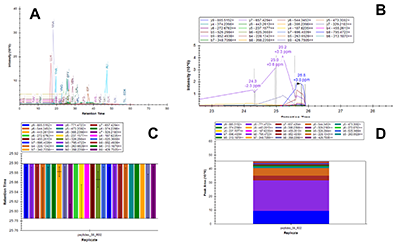
Наиболее критическими факторами, определяющими анализ пептидов в опорной библиотеке, являются: присутствие сигнала детекции пептида; регистрация не менее 50% теоретических фрагментов в тандемном спектре пептида; наличие среди зарегистрированных фрагментов тех, в состав которых могут войти аминокислоты со стабильными тяжёлыми изотопами углерода и азота; желательно, максимальная удалённость пептидов друг от друга по шкале времени удержания на колонке. Для каждого такого пептида отбирается ион с зарядовым состоянием и с максимальной интенсивностью. В отобранном для каждого пептида m/z отбираются не менее 15 фрагментных ионов, ранжированных по интенсивности. Предпочтение отдаётся ионам y-типа, с z=1+ и m/z>450 Th. Таким образом, после всех предпринятых шагов по фильтрации пептидов, опорная библиотека спектров преобразовывается в первичную библиотеку для начала разработки SRM метода (см. X13_56_light_primary_lib). Первичная библиотека. Первичный список транзиций. Первичный SRM анализ. В первичной библиотеке (см. X13_56_light_primary_lib и transitions_56X13_0.xlsx) для всех отобранных пептидов по всем выбранным зарядным состояниям и всем выбранным для них фрагментам проводится проверка детекции сигнала в режиме мониторинга реакций формирования множественных фрагментных ионов (в режиме SRM метода). В том случае, если какой-либо из пептидов, зарегистрированных ранее на этапе тандемного анализа и составления опорной библиотеки, не был зарегистрирован в режиме SRM, проводится обращение к первичным данным опорной библиотеки и повторный выбор и верификация данных на предмет возможного ложного положительного результата. При составлении и доработке первичной библиотеки спектров по результатам SRM анализа для каждого пептидного m/z отбираются не более пяти фрагментных иона с максимальной интенсивностью. На данном этапе разработки метода выбор наиболее предпочтительного пептида в группе белка пока не проводится, так как этап первичной библиотеки необходим только для снижения комплексности набора фрагментных ионов и набора зарядных состояний пептидного иона. Эти шаги невозможно было осуществить на этапе реализации опорной библиотеки, так как спектральныe данные тандемных спектров и SRM-спектров отличаются друг от друга по типу фрагментации (HCD или CID/CAD), геометрии ячейки соударения, нормированной энергии соударения, давлению газа в ячейке соударения и другим параметрам. Таким образом, из первичной библиотеки мы составляем первичный список транзиций (transitions_56X13_1.xlsx), который редуцирован до одного или двух зарядовых состояний ионов каждого пептида и до пяти фрагментов для каждого прекурсора. Вторичная библиотека. Вторичный список транзиций. Вторичный SRM анализ. Весь первичный список пептидов разбивается на кластеры по 60-80 транзиций. При этом не допускается разнесение по разным кластерам транзиций, принадлежащих одному и тому же пептидному иону. Каждый файл данных первичного списка анализируется для составления вторичной библиотеки спектров (см. X13_56_light_secondary_lib). Для составления вторичной библиотеки используют следующие критерии: отбирается только одно m/z пептида. Выбор m/z пептида проводится на основании максимальной интенсивности, отсутствия ложно положительных сигналов по шкале времени удержания, отсутствие интерферирующих сигналов фрагментов. Для выбранного зарядного состояния пептидного иона оставляют три из пяти фрагментных иона, которые обладают максимальной интенсивностью и не интерферируют со сторонним шумовым сигналом. Выбор пептида внутри группы белка на данном этапе также не проводится. Во вторичной библиотеке происходит редукция зарядных состояний и уменьшением потенциальных фрагментных ионов. Полученные данные преобразуем во вторичный список транзиций (см. transitions_56X13_2.xlsx), который не привязан ко времени удержания пептида на колонке, и который разбивается на 2-3 кластера, соответствующих двум-трём методам SRM. Каждый метод содержит в себе набор пептидов, при этом разбиение пептидов одного белка по методам не допускается, также не допускается разбиение фрагментов одной группы прекурсорного иона. Эти условия необходимо соблюдать для сравнения пептидов одного белка для последующего выбора. Основная библиотека. Валидирующий SRM анализ. Валидирующая библиотека Полученные после составления вторичного списка транзиций данные проверяем по следующим критериям: все отобранные пептиды зарегистрированы, сохранение относительных интенсивностей (порядка) транзиций в ходе технических повторов, геометрическая сходимость профилей транзиций, соответствие времени удержания коэффициенту гидрофобности. Если регистрируемый сигнал отвечает критериям выбора, то для каждого пептида фиксируем его время удержания и возможное отклонение по двум техническим повторениям с учётом ширины хроматографического пика для того, чтобы установить вершину и границы окна времени удержания. На данном этапе развития SRM метода мы отбираем один пептид для белка, который максимально отвечает всем требованиям, описанным выше (см. transitions_56X13_3.xlsx). Таким образом, вторичный список транзиций из вторичной библиотеки после анализа данных преобразуется в следующий набор данных: на каждый белок имеется один отобранный пептид, на отобранный пептид имеется одно зарядовое состояние, на отобранное m/z имеется три фрагментных иона. Для каждого пептида фиксируется время удержания на колонке. Набор перечисленных данных преобразуется в валидирующую библиотеку (см. X13_56_light_fundamental_lib) и формирует один метод динамического валидирующего SRM с привязкой ко времени удержания на колонке для каждого пептида (см. transitions_56X13_3.xlsx). Задача данной библиотеки заключается в выборе и последовательной проверке для каждого пептида хроматографических и масс-спеткрометрических характеристик для дальнейшей их оптимизации. Оптимизирующий SRM анализ. Оптимизирующая библиотека После проверки и верификации всех хроматографических и масс-спектрометрических характеристик на основе валидирующей библиотеки составляется промежуточная оптимизирующая библиотека и оптимизирующие методы (см. X13_56_light_optimizing_lib_ed и transitions_56X13_4.xlsx). Оптимизации подлежат такие параметры, как энергия соударения и потенциал ускорения из ячейки соударения. При этом первым оптимизацию проходит параметр энергии соударения. Оптимизация осуществляется подбором энергии соударения в пределах ±20% от расчётной величины для прекурсорного иона. Для каждого прекурсора выбирается та величина энергии соударения, которая на выходе позволяет регистрировать сигнал фрагментов с максимальной интенсивностью (рис. 4).
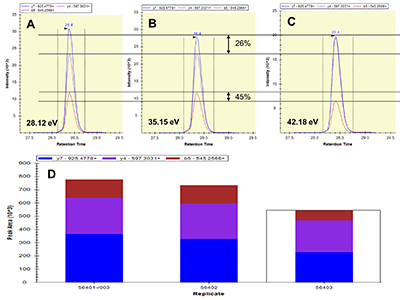
Внутри группы транзиций одного и того же пептидного иона величина энергии соударения, как правило, имеет одинаковую эффективную величину для всех фрагментов, так как данный параметр влияет на эффективность разрушения пептидного иона на этапе первый квадруполь-ячейка. Однако встречаются случаи, когда внутри группы эффективная энергия соударения может отличаться и имеет различную величину для различных фрагментов одного и того же пептида. На втором этапе оптимизации проводится подбор параметра ускоряющего потенциала на выходе из ячейки соударения. Данный параметр оказывает влияние на стабильность и интенсивность фрагментных ионов, сформированных в ячейке соударения после фрагментации, то есть на этапе ячейка соударения - третий квадруполь. В теории, существует обратная зависимость между номинальной масс-зарядной величиной фрагментного иона (без привязки к типу иона и его зарядному состоянию) и значением ускоряющего потенциала, однако на практике зависимость имеет множество исключений и является в большей степени нелинейной. В связи с этим величина ускоряющего потенциала являются более чувствительной и индивидуальной для каждого фрагмента внутри одной группы прекурсорного иона. Параметр ускоряющего потенциала оптимизируется в пределах от 1,5 до 5 В с шагом в 1-1,5 В (рис. 5).
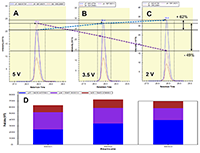
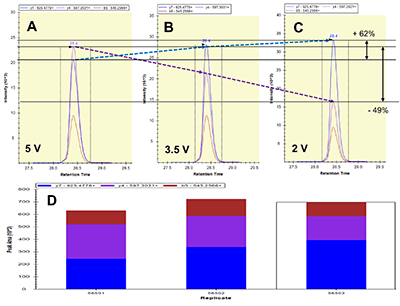
Выбор оптимальной для фрагментного иона величины ускоряющего потенциала при фиксированной величине энергии соударения пептидного иона осуществляется также на основании критерия стабильности, воспроизводимости и максимальной при данной величине интенсивности фрагментного иона. Таким образом, после оптимизации формируется финальная библиотека спектров (см. X13_56_light_final_lib) с финальными приборными параметрами (transitions_final_56X13.xlsx) и масс-спектрометрическими и хроматографическими характеристиками (см. Acq_Method_synth_56.xls). Результирующий SRM анализ. Окончательная библиотека Результирующий метод формируется (56X13_dSRM_Final.m) как динамический метод SRM с окончательно подобранными параметрами и характеристиками с минимальным окном изоляции прекурсорного и фрагментного иона (+/-0.35 а.е.м.), минимальной шириной регистрационного окна времени удержания на колонке (+/- 21-24 с), которое должно отвечать требованию минимального числа конкурирующих в пределах окна времени регистрации ионов (не более 12%), минимального общего времени цикла сканирования (не более 1300 мс) и максимального времени накопления на каждый фрагмент в пределах указанного регистрационного окна времени (не менее 11 мс). Финальным методом смесь пептидов проверяется в нескольких технических повторениях для окончательной проверки воспроизводимости сигнала. Финальный метод формируется отдельно на каждую серию синтеза пептидов (рис. 6).
По итогам окончательных результатов финального метода формируется список пептидов (см. synth_56X13_best_peptides.xlsx) для синтеза стандартных пептидов с включением аминокислотных остатков со стабильными изотопами углерода и азота. Серия финальных методов каждой серии пептидов, приуроченных к выполнению одной и той же задачи, объединяются в один комплексный метод динамического SRM для анализа нескольких сотен пептидов и белков. При необходимости при формировании комплексного метода проводится оптимизации хроматографических условий разделения пептидов на колонке с целью распределения пептидов по оси времени удержания на максимально возможную удалённость их друг от друга за минимально возможное эффективное время общего анализа. Такая оптимизация положительно влияет на величину общего времени цикла сканирования (минимизирует данный параметр) и дискретное время накопления фрагмента (увеличивает данный параметр) при фиксированной ширине регистрационного окна времени удержания. БЛАГОДАРНОСТИ Работа выполнена в рамках программы фундаментальных исследований президиума РАН «Фундаментальные исследования для биомедицинских технологий» на 2018 год. ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi.org/10.18097/BMCRM00006) на сайте журнала. ЛИТЕРАТУРА
|