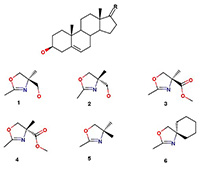
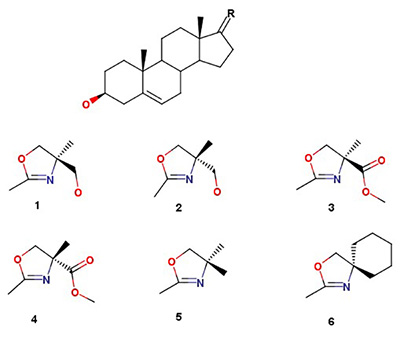
|
Исследование in silico взаимодействия производных 2’-{[(e)-андрост-5-ен-17-илиден]-метил}оксазолиновс андрогеновым рецептором 1Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121 Москва, ул. Погодинская, 10 стр. 8, *e-mail: kirill.soff@gmail.com 2Центр системной биомедицины и биотехнологий, Сколковский институт науки и технологий, Москваa 3Институт физиологически активных веществ Российской академии наук, Московская область, Черноголовка Ключевые слова: рак простаты, андрогеновый рецептор, производные [17(20)E]-прегнена, докинг, молекулярная динамика, метод MM-GBSA DOI: 10.18097/BMCRM00008 ВВЕДЕНИЕ Возникновение и развитие рака предстательной железы (РПЖ) связано с резким повышением уровня андрогенов в простате и гиперактивностью андрогенового рецептора (AR). Современная стратегия терапии РПЖ основана на ингибировании синтеза андрогенных гормонов или подавлении активности AR [1]. В настоящее время в мире широко проводится поиск новых ингибиторов ключевых ферментов биосинтеза андрогенов (в первую очередь ингибиторов стероид-17a-гидроксилазы-17,20-лиазы, CYP17А1) [1-5] и антагонистов AR [6, 7]. Экспериментальные и клинические исследования известных ингибиторов CYP17А1: 17-(3-пиридил)-андрост-5-ен-3β-ола (абиратерона, допущенного для использования в качестве лекарства против РПЖ [8], а также 17-(1H-бензимидазол-1-ил)-андрост-5-ен-3β-ола (галетерона, находящегося на третьей стадии клинических испытаний [9, 10]), показали, что вышеуказанные соединения имеют сродство и к другим клеточным мишеням, в частности к AR, вызывая его инактивацию и/или ускоряя его протеосомную деградацию [9, 11, 12]. Синтезированные ранее азотсодержащие производные [17(20)E]-прегнена [13] ингибировали каталитическую активность СYP17А1 не менее эффективно, чем абиратерон [14, 15], и подавляли рост клеток карциномы простаты линий LNCaP и РС c активностью, сравнимой с таковой для галетерона [15, 16]. Принимая во внимание структурное сходство оксазолиновых производных [17(20)E]-прегнена с абиратероном и галетероном, исследование сродства этих соединений к AR, несомненно, представляет интерес. Целью данной работы является изучение взаимодействия 6 синтезированных ранее оксазолиновых производных [17(20)Е]-прегна-5,17(20)-диена 1–6 c лиганд-связывающим доменом AR методами in silico .МАТЕРИАЛЫ И МЕТОДЫ В работе использовали шесть оксазолиновых производных стероидов, синтезированных в Институте биомедицинской химии им. В.Н.Ореховича [17] (рис. 1). Все соединения были синтезированы в виде Е-изомеров. Однако, было показано, что при взаимодействии с кислыми группами белков данные соединения способны изомеризоваться в Z-конфигурацию [14], в связи с чем эти соединения в работе использовались также и в виде Z-изомеров.
Модели соединений 1-6, были созданы в пакете HyperChem 7.5. Первоначальную оптимизацию структур производили посредством молекулярной механики с использованием силового поля AMBER в пакете HyperChem 7.5 [18]. На втором этапе оптимизацию осуществляли в пакете MOPAC2012 [19] с применением полуэмпирической параметризации РМ7.
Оптимизированные модели лигандов использовали в последующей процедуре молекулярного докинга в программе VinaAutodock [20]. Подготовку лигандов к докингу происходила в пакете AutoDock Tools [21]. В ходе этой процедуры рассчитаны частичные атомные заряды с использованием метода Гастайгера-Хюккеля, определены вращающиеся и невращающиеся связи. В качестве мишени для докинга была выбрана структура лиганд-связывающего домена человеческого андрогенового рецептора в комплексе с синтетическим агонистом EM-5744 (код PDB 2PNU) [22]. Выбор структуры был обусловлен тем, что, несмотря на агонистические свойства EM-5744, данный лиганд имеет стероидный скелет и обладает массивным заместителем в 19 положении. Докинг производили направленно в место связывания природных лигандов в лиганд-связывающий домен рецептора. Область поиска была ограничена решёткой с длиной грани в 20 Å. Решётка была отцентрирована по природному лиганду EM-5744. За правильное расположение лиганда принимали конформации, для которых среднеквадратичное отклонение (RMSD) не превышало 1,5 A по сравнению со стероидным скелетом EM-5744. Анализ результатов докинга осуществлялся при помощи программы LigPlot+ [23]. Оптимизацию положения лигандов проводили методом моделирования молекулярной динамики. Расчёты молекулярной динамики (МД) проводили при помощи пакета программ AMBER 9.0 [24] в явно заданном растворителе (TIP3P) с использованием периодических граничных условий. Параметризацию атомов белка проводили с применением силового поля amber99-SB, лигандов - силового поля GAFF. Для нейтрализации системы использовали ионы Cl-. На первом этапе проводили последовательную минимизацию системы в вакууме и в растворителе на протяжении 25000 шагов с применением метода градиентного спуска. На следующем этапе были последовательно произведёны нагрев системы до 300 К и повышение давления с применением NVT и NPT ансамблей. Молекулярную динамику проводили на траектории 15 нс с шагом 2 фс. Расчет электростатических взаимодействий проводили с применением метода PME (Particle Mesh Ewald) на расстоянии до 8.0Å. Поддержание заданных параметров температуры и давления осуществляли с применением термостата Ланжевена и баростата Берендсена.
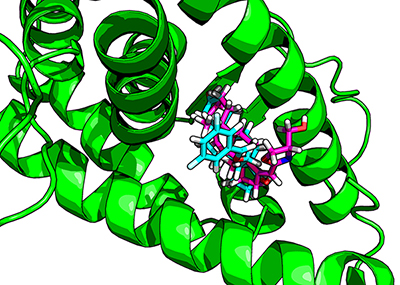
Анализ траекторий МД проводили при помощи пакета VMD [25]. Энергия связывания лигандов c белком рассчитывали методом MM-GBSA [26]. Расчет энергии проводили на временном отрезке 9-15 нc на основе мгновенных состояний системы. Электростатический вклад в энергию связывания рассчитывали c использованием Generalized Born метода. Все вычисления были произведены на вычислительном сервере ИБМХ (HP ProLiant G8, AMD Opteron 6176 SE *2, (48 threads), 64GB RAM, x8 2.5" SAS/SATA с применением ускорителях Nvidia Tesla). РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Первоначально докинг соединений 1-6 в Z- и E-конформациях был проведен в полноразмерную модель лиганд-связывающего домена AR. Однако, решений докинга ни для одного соединения получено не было. В связи с этим было предположено, что поскольку для докинга была использована структура AR в комплексе с агонистом, то расположение в ней спирали Н12 (закрывающей вход в место связывания) может препятствовать корректному размещению лигандов в лиганд-связывающем кармане. Считается, что при связывании агониста в лиганд-связывающем домене спираль Н12 закрывает вход в место связывания и участвует в формировании места связывания для активаторного пептида, тогда как антагонисты препятствуют правильному расположению этой спирали, в результате активаторный пептид не может связаться с рецептором [27]. Для проверки предположения эта спираль была удалена. Ранее подобный прием был использован в ряде работ [28, 29]. Повторный докинг всех соединений в лиганд-связывающий домен с отсутствующей спиралью Н12 позволил получить решения докинга для всех соединений. Все позы были получены только для соединений 1-6 в Z-конформациях. Положение стероидных ядер у исследуемых молекул хорошо совпадало с положением молекулы EM-5744 из кристаллической структуры. (Рис. 2).
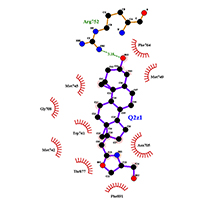
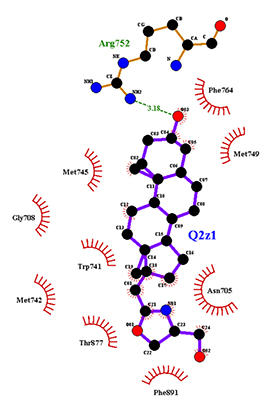
На рисунке 3 представлены контакты соединения 2, определенные с помощью программы LigPlot+.
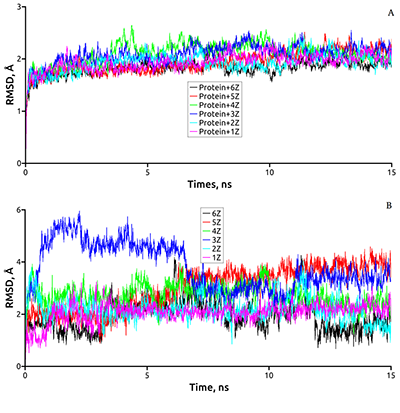
Оценка стабильности полученных комплексов соединений 1–6 с лиганд-свзязывающим доменом AR была проведена методом молекулярной динамики (рис. 4). Анализ значений среднеквадратичных отклонений атомов аминокислотных остатков (RMSD) показал, что структуры всех исследуемых комплексов оставались стабильны (рис. 4,А). Значения RMSD выходили на “плато” после 2 нс и в среднем составляли около 2 Å. Анализ RMSD для самих лигандов в комплексе с AR на траектории МД показал, что наиболее устойчивыми оставались положения соединений 1, 2, 4 и 2 (Рис. 4, Б). Их отклонение от первоначального положения составляло 2-3 Å. Наибольшее отклонение наблюдалось для соединения 3. Сначала величина RMSD для него возрастала до 6 Å, потом постепенно снизилась и в дальнейшем колебалась в районе 3-4 Å. То есть этот комплекс был наименее устойчив в ходе динамики. Визуальная оценка изменения положений лигандов в ходе МД выявила, что наибольшему смещению относительно начальных позиций подверглись лиганды 3 и 5, что отражается в высоком значении их RMSD.
Оценка способности соединений 1–6 образовывать водородные связи с белком в ходе молекулярной динамики показала, что у большинства лигандов водородная связь с белком присутствовала на протяжении не более 20% от общего времени динамики. Лишь соединение 5 образовывало водородную связь на протяжении 53% времени. На основе траекторий молекулярной динамики были рассчитаны значения энергий связывания соединений 1–6 с лиганд-связывающим доменом AR методом MM-GBSA (таблица 1). Расчет энергий проводился на участке траектории 9-15 нс, когда значения RMSD систем уже вышли на плато. Наибольшей энергией связывания характеризуются соединения 5 (-57.9 ккал/моль) и 6 (-57.1 ккал/моль).
Докинг производных 17(20)Е-прегна-5,17(20)-диена (соединения 1-6) в AR удался лишь в мишень, в которой отсутствует спираль Н12. Заместители у атома С17 при этом располагались в месте, которое занимает Н12-спираль AR при его связывании с агонистами. Это может указывать, что данные соединения при связывании с AR должны препятствовать связыванию этой спирали с основной глобулой домена и таким образом проявлять антагонистические свойства. Все варианты докинга были получены для соединений в Z-конформации, что предполагает, что, как и в случае с взаимодействием этих соединений с цитохромом Р450 17А1 [14], предварительно должна произойти кислотная изомеризация этих соединений. Результаты докинга показали, что все соединения могут разместиться в месте связывания таким образом, что их гидроксильная группа при С3-атоме стероидного кольца образует Н-связь с Arg752 белка, что также характерно для других стероидных лигандов ядерных рецепторов. Все описанные соединения в ходе МД сохраняют указанную выше водородную связь. Эта водородная связь, за исключением соединения 5, присутствовала только 20% времени. Для соединения 5 эта величина составляла 50%. Ранее эти соединения были исследованы на способность ингибировать активность цитохрома Р450 17А1, и было показано, что часть из них обладает низкой ингибиторной способностью [30]. Так, соединение 5 было на порядок менее активно, чем абиратерон (лекарство, в основном действующее на цитохром Р450 17А1), а соединение 6 было абсолютно неактивно. Таким образом, проведенные исследования показали, что некоторые азотсодержащие производные [17(20)E]-прегнена способны связываться с AR, причем соединения 5 и 6 являются вероятными антагонистами AR и могут быть рекомендованы для экспериментальной проверки на антиандрогенное действие. БЛАГОДАРНОСТИ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. ЛИТЕРАТУРА
|