|
Предсказание аффинности прогестинов к ядерному рецептору прогестерона на основе данных с коррекцией RBA Научно-исследовательский институт биомедицинской химии имени В. Н. Ореховича, 119121, Москва, Погодинская ул. 10;*e-mail: a.v.mikurova@ibmc.msk.ru Ключевые слова: рецептор прогестерона; аффинность; молекулярный докинг; молекулярная динамика; MM-PBSA DOI: 10.18097/BMCRM00080 ВВЕДЕНИЕ
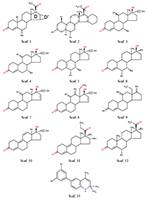
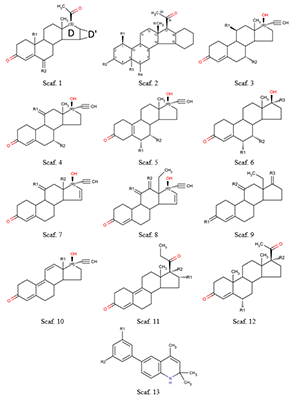
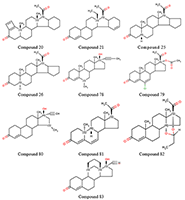
В настоящее время опубликовано большое число работ, посвящённых оценке in silico афинности как стероидных, так и нестероидных прогестинов к рецептору прогестерона [1] Данные препараты широко применяются в контрацепции, заместительной и противоопухолевой терапии [2,3]. Однако большое число таких работ, демонстрирующих неплохие результаты, ограничиваются гомологичным рядом химических соединений [4-6] и чаще всего используют технологию CoMFA [7], a priory нацеленную на анализ высокогомологичных рядов. В отличие от CoMFA, технологии, использующие такие процедуры как молекулярный докинг, молекулярную динамику, MM-PBSA (MM-GBSA) [8] или другие способы анализа модельных комплексов, позволяют делать предсказания на моделях, обученных на одном гомологичном ряде химических соединений для рядов, отличных от использованных в обучении соединений. Ранее мы уже успешно применяли процедуры молекулярных динамики и докинга для создания уравнений предсказания аффиности производных 16α,17α-циклоалканопрогестерона к рецептору прогестерона (RP) [6]. Однако тогда был использован ряд соединений, представляющий собой группу стероидов, имеющих несущественные структурные различия. В настоящей работе число стероидных соединений было расширено. Кроме того была добавлена небольшая группа соединений нестероидной природы. МАТЕРИАЛЫ И МЕТОДЫ Структуры химических соединений и активность в отношении ядерного рецептора прогестерона [6,9,10] представлены на рисунках 1-2 и в таблице 1. Следует отметить, что в различных источниках авторы используют неодинаковые параметры в качестве оценки афинности либо степени ингибирования: в двух выборках используется относительная связывающая активность (Relative Binding Affinity, RBA), а в третьей – IC50. Кроме того, референсное соединение для разных выборок также различается. В нашем случае это либо прогестерон [6], либо мифепристон [10], либо соединение 74 [9]. RBA прогестерона по отношению к мифепристону ниже в 2 раза [6], а соединения 74 в 7 раз [9]. Эти соотношения были использованы для пересчета значений и приведения данных к единому референсному соединению. С учетом применения авторами [6,9,10] различных способов оценки биологической активности соединений, наиболее адекватным представляется использовать непосредственно величину RBA и расчётную величину RIA (Relative Inhibition Activity) относительно единого соединения (прогестерона). Для построения уравнений использовали десятичный логарифм RBA (RIA), что позволяет регуляризовать ошибку предсказания. Разумеется, напрямую объединять величины RBA и RIA в одну выборку нельзя, однако можно ожидать, что данные величины должны между собой хорошо коррелировать, так как в основе своей имеют одно и тоже явление – связывание с рецептором. Учитывая, что выборка с RIA мала, её использовали как тестовую.
Не все данные были получены по отношению к рецептору прогестерона человека. Данные, взятые из работы [6], получены на рецепторе кролика. Возможность использовать эти данные для анализа с использованием рецептора человека обсуждается в работе [6] и основана на высокой степени идентичности RP у этих видов и отдалённости имеющихся аминокислотных различий от сайта связывания лиганда.
Все структуры лигандов были построены и оптимизированы средствами программы Sybyl-X [11] с расстановкой частичных атомных зарядов по эмпирической схеме с использованием поля сил MMFF [12]. Эту же схему частичных атомных зарядов использовали для лигандов и на последующих шагах.
Моделирование комплексов осуществляли посредством докинга молекул потенциальных лигандов к участку связывания прогестерона в лиганд-связывающем домене (Ligand Binding Domain, LBD) RP. Структура LBD была взята из файла 1A28 [13] в Protein Data Bank. Построение моделей предсказания аффинности проводили на базе параметров комплексов, рассчитанных методами MM-PBSA и MM-GBSA [8]. Процедуры докирования и молекулярной динамики выполняли средствами программ Dock 6.7 [14] и Amber 16.0 [15] (поля сил FF14SB и GAFF2) по схеме, примененной ранее [16]. Для выполнения расчетов был использован гибридный высокопроизводительный вычислительный комплекс Федерального исследовательского центра «Информатика и управление» Российской Академии Наук (ФИЦ ИУ РАН) [17] IBM Minsky (2xPower8, 4хTesla P100). Основной объём вычислений проводили с использованием GPU ускорителей. Часть вычислений выполняли на CPU, так как использование GPU было либо неэффективно, либо не поддерживалось для данного типа вычислений. Среднее время расчётов на 1 комплекс составило 34 мин для вычислений на GPU и 5 ч на CPU. Этапы вычислений:
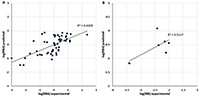
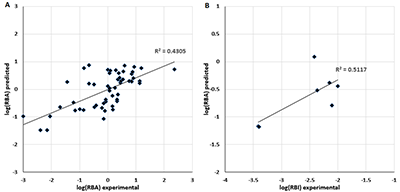
Рассматривали два способа отбора вариантов. Первый – по усреднённому по 25 состояниям значению изменения свободной энергии комплекса, рассчитанному по методу MM-PBSA. Второй (для стероидов) – отбор решения, подобного расположению прогестерона в кристаллической структуре. Значения отдельных энергетических компонентов, рассчитанных методом MM-PBSA/MM-GBSA, использовали в качестве независимых переменных для уравнений предсказания аффинности. Были использованы: изменение величины электростатического взаимодействия (ELE); изменение величины ван-дер-ваальсовых взаимодействий (VDW); гидрофобный (PBSUR) и сольватационный (PBCAL) вклады в изменение свободной энергии, рассчитанной методом Пуассона-Больцмана (PB); аналогичные вклады, рассчитанные обобщенным методом Борна (GB) (GBSUR и GB); рассчитанные модулем NMODE [9] значения трансляционного (TSTRA), ротационного (TSROT) и колебательного (TSVIB) энтропийных вкладов; молекулярный вес лиганда. Уравнения линейной регрессии оценивали по величине Q2 в процедуре скользящего контроля методом исключения по одному. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Результаты процедуры докирования отражены в таблице 1. Не для каждого случая программа Dock нашла решение. Кроме того, как видно из таблицы 1, не в каждом случае лучшим по изменению энергии рассчитанной методом MM-PBSA было положение, аналогичное положению прогестерона в кристалле. Для нестероидных соединений у нас не было априорного положения, поэтому в качестве основного была отобрана та ориентация лиганда, которая чаще всего была лучшей согласно энергетической оценке (рис. 2). В таблице 2 представлены статистические характеристики ряда уравнений, полученных для двух отдельных выборок со стероидными соединениями и объединённой выборки, а также результаты тестирования на выборке нестероидных соединений. Наилучшие результаты в обучении показала выборка 1 (R2обучения 0.73). Однако, учитывая, что в обучении использовали только 23 соединения, уравнение с 9 переменными может быть не совсем адекватным. Это видно на примере тестовой выборки (R2предсказания 0.04) и выборки 2 (R2предсказания 0.1). В данных случаях R2тестирования значительно хуже, чем R2обучения. Под R2тестирования для тестовой выборки понимается величина корреляции (R2) между предсказанной величиной RBA и наблюдаемой величиной RBI. Выборка 2 показала весьма посредственный результат (R2обучения 0.16). Соответственно, тестирование на выборке 1 и тестовой выборке также демонстрирует неадекватный результат. Объединение выборки позволяет, с одной стороны, увеличить статистическую значимость полученного уравнения (R2обучения 0.44 – 0.47), а с другой – получить вполне удовлетворительные результаты (рис. 3) для предсказания величины RBI тестовой выборки (R2тестирования 0.51). Модель позволяет уверено ранжировать соединения тестовой выборки по прочности связывания с RP.
ЗАКЛЮЧЕНИЕ Таким образом, можно заключить, что при использовании достаточно длительной процедуры молекулярной динамики параметры, полученные в результате применения процедуры MM-PBSA (MM-GBSA), можно использовать для создания уравнений предсказания аффинности, причём использование величины RBA позволяет объединить в одну обучающую выборку данные, полученные в различных условиях, а также оценить величины, отличные от RBA. БЛАГОДАРНОСТИ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. Подготовка и отладка программного обеспечения выполнялась при поддержке гранта РФФИ № 18-29-03100. ЛИТЕРАТУРА
|