|
СОДЕРЖАНИЕ 1. ТЕХНОЛОГИИ ОДНОМОЛЕКУЛЯРНОГО СЕКВЕНИРОВАНИЯ 1.2. Нанопоровое секвенирование 2. ИЗУЧЕНИЕ ТРАНСКРИПТОМА С ПОМОЩЬЮ ТЕХНОЛОГИЙ ОДНОМОЛЕКУЛЯРНОГО СЕКВЕНИРОВАНИЯ 2.1. Применение SMRT-секвенирования в транскриптомном анализе 2.2. Анализ транскриптома с использованием нанопорового секвенирования |
Перспективы использования секвенаторов третьего поколения для количественного профилирования транскриптома
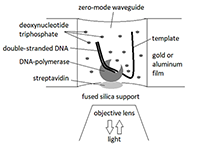
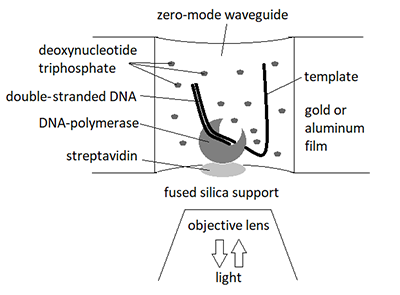
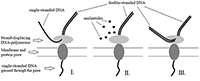
1Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича, Ключевые слова: секвенирование третьего поколения; транскриптом; количественное профилирование DOI: 10.18097/BMCRM00086 ВВЕДЕНИЕ Инициирование и успешное выполнение проекта «Геном человека» дало начало новым, так называемым «омиксным» подходам к исследованию фундаментальных и прикладных аспектов функционирования живых существ. Характерной чертой этих подходов является получение и биоинформатический анализ огромных массивов данных [1]. Дополнительно к геномике, сегодня окончательно сложились и активно развиваются такие «омиксные» направления, как протеомика, транскриптомика, метаболомика и интерактомика, каждый из которых опирается на свои технологические платформы [1]. Транскриптомика, которая в первую очередь фокусируется на идентификации транскриптов во всем разнообразии их изоформ и на количественной оценке их представленности в разнообразных типах клеток (количественное профилирование транскриптома), опиралась в начале своего развития на технологию микрочипов, первоначально разработанную для геномного анализа [2]. В настоящее время использование технологий секвенирования, известных как «секвенирование нового поколения» (Next Generation Sequencing, NGS), стало доминирующим методологическим подходом к анализу транскриптома, включая его количественное профилирование [3]. Сегодня NGS подразделяют на секвенирование второго и третьего поколений (SGS и TGS – Second and Third Generation Sequencing) [4], технологии которых существенно различаются. Одним из характерных различий SGS и TGS является размер секвенируемых молекул. В случае SGS секвенируются относительно небольшие (от 25 до 500 пар оснований (п.о.)) фрагменты нуклеиновых кислот с перекрывающимися последовательностями. По аналогии с принятым в англоязычной литературе термином «reads», последовательности ДНК таких фрагментов в русскоязычной литературе называют «чтениями». После завершения этапа секвенирования, «чтения» собираются в протяженные участки генома с помощью специальных математических алгоритмов (aligners), позволяющих проводить их выравнивание [5]. При транскриптомном анализе проводят секвенирование фрагментов кДНК, которые потом собирают в последовательности транскриптов путём их выравнивания с использованием референсных геномов или транскриптомов (картирование), или без их использования (секвенирование транскриптома de novo). При количественном профилировании транскриптомов методами SGS в качестве метрики используют нормированное специальным образом количество «чтений», картирующихся на определённый ген (транскрипт): RPKM (Reads Per Kilobase Per Million mapped reads) или FPKM (Fragments Per Kilobase Per Million mapped reads), что позволяет определять относительную представленность транскриптов в исследуемом образце [6]. В случае TGS длина «чтения» может достигать десятков тысяч п.о. [7]. Применительно к транскриптому такая длина «чтения» делает возможным проведение так называемого «одномолекулярного секвенирования» (single molecule sequencing), когда практически любой транскрипт секвенируется как единичная молекула, что устраняет необходимость последующей «сборки». Таким образом, секвенаторы третьего поколения можно рассматривать как потенциальные молекулярные счётчики, позволяющие определять представленность каждого транскрипта в транскриптоме с высокой производительностью, чувствительностью и специфичностью. В данном обзоре рассмотрены основные технологические особенности одномолекулярного секвенирования с использованием технологических платформ, разработанных компаниями «Pacific Biosciences» (США) и «Oxford Nanopore Technologies» (Великобритания), а также их применение в транскриптомном анализе, включая количественное профилирования транскриптома. 1. ТЕХНОЛОГИИ ОДНОМОЛЕКУЛЯРНОГО СЕКВЕНИРОВАНИЯ Термин «одномолекулярное секвенирование» первоначально появился как часть названия технологии секвенирования ДНК, разработанной компанией «Pacific Biosciences» и получившей название «одномолекулярное секвенирование в реальном времени» (Single Molecule Real Time sequencing – SMRT) [8]. В дальнейшем он был применён для обозначения технологии нанопорового секвенирования, предложенной компанией «Oxford Nanopore Technologies» , также характеризующегося длиной «чтения», достигающей десятки тысяч п.о. [9,10]. 1.1. SMRT-секвенирование Технология SMRT-секвенирования основана на наблюдении в реальном времени за синтезом ДНК-полимеразой новой цепи на ДНК-матрице, которая и представляет секвенируемую молекулу ДНК [11,12]. Иллюстрация концепции SMRT-секвенирования представлена на рисунке 1. Технология включает два принципиальных компонента: 1) плоский световод, который позволяет направлять световую энергию в ячейку, размер которой значительно меньше длины волны видимого света (zero-mode waveguide, ZMW); 2) дезоксинуклеозидтрифосфаты (dNTP), у которых к концевой фосфатной группе присоединен флуорофор.
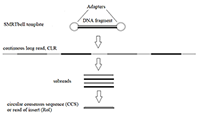
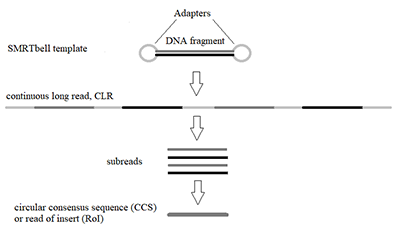
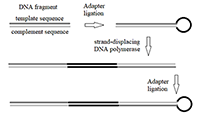
ZMW-ячейка представляет подложку из плавленого кварца, покрытую металлической плёнкой (алюминиевой или золотой) толщиной ~100 нм, в которой сделано отверстие диаметром ~100 нм. Особенность распространения света в отверстии с такой апертурой состоит в том, что его интенсивность быстро затухает, в результате чего освещённой оказывается лишь малая часть ячейки (объемом около 20 зептолитров), прилегающая к поверхности подложки [4]. Время диффузии флуоресцентно-меченных dNTP (далее фм-dNTP) через такой малый объем составляет несколько микросекунд. Дно ячейки дериватизировано полиэтиленгликолем (ПЭГ), конъюгированным с биотином. В качестве ДНК-полимеразы используется мутантный вариант рекомбинантной ДНК-полимеразы бактериофага φ29 с пониженной 3’-5’ экзонуклеазной активностью, биотинилированный in vivo на N-конце [13]. После формирования комплексов ДНК-полимеразы с ДНК-матрицей, к ним добавляется стрептавидин, что позволяет иммобилизовать комплекс полимераза-матрица на дне ячейки через формирование мультимерного комплекса ПЭГ-стрептавидин-полимераза-матрица [12]. Присоединение флуорофора к концевой фосфатной группе dNTP (в отличие от более распространённого подхода к его конъюгации с dNTP через присоединение к основанию) приводит к отщеплению флуорофора полимеразой при включении нуклеотида в синтезируемую цепь ДНК. В результате нуклеотиды в синтезируемой цепи не мечены флуорофорами и не дают вклада в флуоресцентный сигнал. Характерное время между захватом фм-dNTP активным центром ДНК-полимеразы и отщеплением флуорофора при включении нуклеотида в растущую цепь ДНК составляет несколько мс, что позволяет дискриминировать такое событие от прохождения фм-dNTP через освещенный объем ячейки в силу броуновского движения, основываясь на длительности сигнала. Каждый тип dNTP мечен флуорофором, характеризующимся своим «цветом» (испускающим свет определенной длины волны), что позволяет наблюдать процесс синтеза в реальном времени как последовательность флуоресцентных сигналов разного «цвета» [4,12]. Регистрация сигнала во времени происходит с помощью опто-электронной системы, сопрягающей возможности конфокальной микроскопии, фото-электронного усиления сигнала и CCD-камеры (CCD – charge-coupled device) и позволяющей детектировать единичные молекулярные события, такие как включение флуоресцентно-меченного dNTP в растущую цепь ДНК [11,12,14]. Характерной особенностью SMRT-технологии является использование в качестве матрицы топологически замкнутой (кольцевой) ДНК (SMRTbell template [15]), которую получают лигированием ДНК-адаптеров, имеющих структуру «шпильки», к фрагменту двунитевой ДНК (рис. 2). ДНК-полимераза фага φ29 является «вытесняющей ДНК-полимеразой» (strand displacing DNA polymerase), т.е. способна вытеснять одну из нитей ДНК при синтезе, в результате чего «чтения» включают как «смысловые», так и «антисмысловые» последовательности секвенируемого фрагмента ДНК, разделённые последовательностями адаптеров и повторяющиеся много раз [15,16]. Результатом является так называемое «непрерывное длинное чтение» (continuous long read, CLR), которое при обработке данных путём узнавания и устранения последовательностей адаптеров может быть преобразовано в набор «субчтений» (subreads). Консенсусная последовательность «субчтений» единичного CLR носит название «циркулярная консенсусная последовательность» (circular consensus sequence, CCS) или «чтение вставки» (read of insert, RoI). Обработка CLR и получение набора RoI происходят в автоматическом режиме с использованием программного обеспечения, предоставляемого «Pacific Biosciences». Биоинформатический инструментарий для анализа результатов SMRT-секвенирования также доступен на GitHub [17].
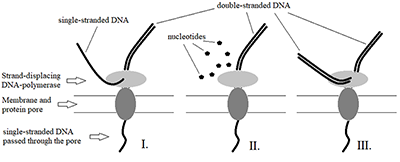
Высокая производительность секвенирования достигается тем, что синтез ДНК проводиться параллельно в тысячах ZMW-ячейках, расположенных с высокой плотностью на плоской кварцевой подложке (ZMW-чип) [12]. Так, в первом секвенаторе третьего поколения PacBio RS, который был выведен компанией «Pacific Biosciences» на рынок в 2011 г., использованы чипы, содержащие 3 тыс. ZMW-ячеек. В 2013 г. компания начала продажи секвенатора PacBio RS II, где параллельное секвенирование проводится в 150 тыс. ZMW-ячейках. Тем не менее, производительность SMRT-секвенирования заметно ниже, чем производительность многих секвенаторов второго поколения – как правило, пропускная способность одного чипа, содержащего 150 тыс. ZMW-ячеек, составляет 0.5 - 1 млрд. нуклеотидов (нт) [16]. Следует отметить также, что фактически только около 35 - 75 тыс. ZMW-ячеек из 150 тыс. содержат одну иммобилизованную молекулу ДНК-полимеразы на ячейку и могут быть использованы для секвенирования (остальные либо не содержат ДНК-полимеразы, либо содержат более одной молекулы полимеразы из-за пуассоновского распределения числа ДНК-полимераз по ZMW-ячейкам при иммобилизации) [12,16]. В настоящее время «Pacific Biosciences» предлагает систему одномолекулярного секвенирования в реальном времени Sequel System (с 2016 г.), использующую чипы, содержащие 1 млн. ZMW-ячеек, что повышает пропускную способность до 3.5 - 7 млрд. нт на ZMW-чип [16]. Одновременно с увеличением пропускной способности происходила оптимизация и совершенствование протоколов секвенирования и используемых реагентов (включая рекомбинантную ДНК-полимеразу), что привело к возрастанию средней длины чтения: в секвенаторах PacBio RS медианное значение длины чтения составляло 4 тыс. нт, в PacBio RS II – около 10 тыс. нт, а в Sequel System – более 20 тыс. нт. Максимальная длина чтения превышает 60 тыс. нт, а в некоторых случаях может даже достигать более 90 тыс. нт [18]. Одним из существенных недостатков SMRT-секвенирования является высокая частота ошибки, которая достигает 15% [4,19,20]. Среди причин столь высокого уровня ошибок – захвать dNTP активным центром ДНК-полимеразы без последующего включения основания в растущую цепь [19]. При этом время нахождения «захваченного» фм-dNTP в освещенном объеме ZMW-ячейки может также составлять мс и восприниматься системой как включение нуклеотида в растущую цепь. Поскольку ошибки распределены случайно по длине «чтения», они могут быть идентифицированы и устранены, если последовательность ДНК-фрагмента повторяется в «чтении» достаточное количество раз (что достигается использованием SMRTbell-матрицы). Так, при 15-кратном повторе вероятность ошибки становиться менее 1% [20], а при 30-кратном точность секвенирования - более 99.999% [18]. Однако по мере увеличения длины секвенируемого ДНК-фрагмента количество повторов в «чтении» в среднем уменьшается, что приводит к возрастанию уровня ошибок [20]. Существенным недостатком SMRT-технологии является то, что иммобилизация комплексов ДНК-полимераза/SMRTbell-матрица в ZMW-ячейке зависит от размера матрицы: комплексы с матрицами меньшего размера иммобилизуются со значительно более высокой эффективностью, что приводит к превалированию коротких ДНК-фрагментов [16]. Применительно к транскриптомному анализу это означает, что короткие транскрипты будут секвенироваться со значительно большей частотой, чем они встречаются в секвенируемой популяции молекул РНК. Для решения данной проблемы «Pacific Biosciences» предлагает проводить предварительное разделение кДНК по размеру на фракции: 1 - 2 тыс. нт, 2 - 3 тыс. нт, 3 - 6 тыс. нт и 5 - 10 тыс. нт. Фракционирование кДНК проводится с помощью либо традиционного агарозного электрофореза, либо, что предпочтительно, гель-электрофореза в пульсирующем поле (pulsed field gel electrophoresis, PFGE) [16]. В последнем случае производитель рекомендует использовать PFGE-системы BluePippin или SageELF компании «Sage Science» (США). Так как процедура фракционирования кДНК по размеру приводит к значительному уменьшению количества материала, что может привести к потере низкокопийных транскриптов, рекомендуется проводить ПЦР-амплификацию кДНК перед фракционированием и каждой фракции после фракционирования перед созданием SMRTbell-библиотеки. Метод транскриптомного анализа, разработанный компанией Pacific Biosciences, включающий получение кДНК(1), пре-амплификацию кДНК (2), фракционирование кДНК по размеру (3), пост-амплификацию кДНК (4), сознание SMRTbell-библиотеки для каждой фракции (5) и её последующее SMRT-секвенирование (6), получил название Iso-Seq [16]. Следует отметить, что технология SMRT может потенциально быть использована для прямого секвенирования молекул РНК, если ДНК-полимеразу заменить на обратную транскриптазу [21]. Используя обратную транскриптазу вируса иммунодефицита человека, авторы [21] показали возможность мониторинга синтеза кДНК на РНК-матрице в ZMW-ячейке в реальном времени. Метод позволил идентифицировать модификации оснований РНК и наблюдать перестройки вторичной структуры молекул РНК. Однако дальнейшего развития применение SMRT-технологии для прямого секвенирования РНК за прошедшие после публикации 5 лет так и не получило. 1.2. Нанопоровое секвенирование Нанопоровое секвенирование нуклеиновых кислот основано на пропускании молекул однонитевой ДНК (онДНК) или РНК через поры диаметром 1 - 2 нм. Появлению нанопоровых секвенаторов предшествовала череда академических исследований, посвящённых анализу прохождения ДНК и РНК через белковые нанопоры, формируемые поринами – специализированными трансмембранными белками бактерий – в липидном бислое. Первое исследование такого рода относится к 1996 г., когда Kasianowicz и соавт. [22] показали, что гомоолигонуклеотиды ДНК под действием приложенного напряжения могут проходить через поры, образуемые α-гемолизином (α-ГЛ) при его встраивании в искусственную липидную мембрану (диаметр такой поры составляет 1.4 нм). При этом происходит временная блокировка поры, которая сопровождается уменьшением количества ионов электролита, проходящих через пору под действием приложенного напряжения и, соответственно, снижением силы тока, протекающего через мембрану. Авторы также показали, что величина снижения силы тока зависела от типа нуклеотидов, образующих гомополимер ДНК, и что днунитевая ДНК (днДНК), представленная дуплексами гомоолигонуклеотидов, была не способна пройти через пору [22]. Позднее они же показали, что прохождение через α-ГЛ-нанопору синтетической РНК, состоящей из двух сегментов, образованных разными нуклеотидами (включая нуклеотиды с метилированными основаниями), может быть детектировано как ступенчатое падение силы тока [23]. Эти работы продемонстрировали принципиальную возможность секвенировать нуклеиновые кислоты, основываясь на детекции последовательности изменений величины силы тока, протекающего через мембрану, при транслокации молекулы онДНК или РНК через встроенную в мембрану белковую пору. Однако практическая реализация этой возможности требовала решения по меньшей мере двух задач [24]: (1) замедления транслокации молекул онДНК и РНК с 100 - 1000 нт в миллисекунду до ~1 нт в миллисекунду, чтобы позволить временное разрешение их прохождения через нанопору на однонуклеотидном уровне, и (2) распознавания последовательностей нуклеотидов, формирующих сигнал (длина поры в α-ГЛ такова, что сигнал генерируется участком последовательности, включающим 12 нт). Возможные решения этих задач были показаны в работах [25-27]. Как оказалось, формирование комплекса нуклеиновой кислоты с моторным белком, в качестве которого была использована ДНК-полимераза фага φ29, может замедлить прохождение онДНК через α-ГЛ-нанопору до 2.5 - 40 нт/с, что позволяет регистрировать её транслокацию с временным разрешением на однонуклеотидном уровне [25,26]. Авторы работ продемонстрировали, что различные активности φ29 ДНК-полимеразы (которые проявляются в зависимости от доступности для полимеразы ионов магния и/или дезоксинуклеотидтрифосфатов и включают геликазную, 3’-5’-экзонуклеазную и полимеразную активности) могут быть использованы для контроля скорости транслокации онДНК через пору (рис.3). Решением второй задачи стала замена α-ГЛ на генетически модифицированный рекомбинантный вариант белка MspA (Mycobacterium smegmatis protein A – трансмембранный белок Mycobacterium smegmatis). В отличие от α-ГЛ, длина поры MspA такова, что сигнал генерируется участком онДНК, состоящим из 4 нт, при этом замена даже одного нуклеотида на другой в таком «квадромере» приводит к детектируемому изменению амплитуды сигнала[27]. Интегрирование возможностей, которые давало одновременное использование φ29 ДНК-полимеразы и «мутантной» MspA-нанопоры, продемонстрировало, что транслокация различных последовательностей ДНК длиной 42-53 нт через нанопору сопровождается своим (т.е. характерным для каждой тестированой последовательности) паттерном изменений силы тока во времени [28]. Экспериментальное исследование характеристических паттернов изменений силы тока для всех 256 возможных четырёхбуквенных комбинаций нуклеотидов dA, dT, dC и dG позволило составить «карту квадромеров», с помощью которой оказалось возможным конвертировать последовательности изменений в силе тока при трансклокации длинных (до 4.5 тыс. нт) фрагментов ДНК фага φX174 в нуклеотидные последовательности, которые однозначно картировались на геном фага [29].
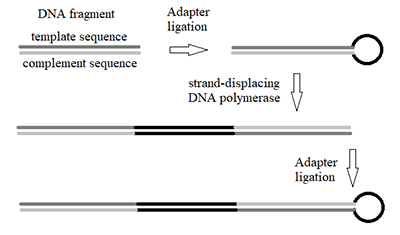
Очевидно, эти же принципы лежат в основе метода нанопорового секвенирования нуклеиновых кислот, разработанного и коммерциализированного компанией «Oxford Nanopore Technologies» («ONT»), хотя она не раскрывает многих деталей устройства и функционирования предлагаемых ею секвенаторов. Тем не менее, в патентном портфеле «ONT» находятся патенты, защищающие её права на генетически модифицированные рекомбинатные α-ГЛ [30], MspA [31] и лизинин (порин, найденный у дождевого червя Eisenia fetida) [32] и на использование геликаз Hel308 [33] и XPD [34] в качестве моторных белков для контролируемой транслокации нуклеиновых кислот через белковую нанопору. Также компанией запатентован метод секвенирования днДНК, который состоит в лигировании ДНК-адаптера, имеющего шпилечную структуру, к одному из концов фрагмента днДНК (который может представлять фрагмент геномной ДНК или дуплексную кДНК), что обеспечивает последовательное «чтение» обоих нитей ДНК при использовании в качестве моторного белка ДНК-полимеразы фага φ29, которая, как оказалось, при определённых условиях способна контролировать транслокацию онДНК через нанопору даже взаимодействуя только с единичной ДНК-цепью [35]. В отличие от нанопорового секвенирования только одной цепи днДНК (так называемое 1D-секвенирование, в этом случае получают так называемое «чтение матрицы» – «template read»), в случае использования такого адаптера происходит «чтение» обоих цепей (2D-секвенирование, включающее «чтение матрицы» и «чтение комплемента» – «complement read»). Это позволяет при дальнейшем анализе данных провести коррекцию ошибок распознавания нуклеотидов, которые распределены случайно в обоих «чтениях», и, таким образом, повысить точность секвенирования. Интересно, что используя вытесняющую ДНК-полимеразу, можно превратить фрагмент днДНК с адаптером в днДНК, в которой последовательности «матрицы» и «комплемента» будут повторяться дважды (рис. 4).
Последующее лигирование адаптера к такой днДНК позволит «прочитать» дважды как «матрицу», так и «комплемент» исходного фрагмента днДНК, ещё более повышая точность нанопорового секвенирования [35]. Компанией «ONT» также запатентованы математический алгоритм распознавания нуклеотидов (base-calling), позволяющий конвертировать последовательность изменений силы тока во времени при трансклокации онДНК через нанопору в последовательность нуклеотидов, который основан на использовании скрытой марковской модели и алгоритма Витерби [36], а так же метод и аппарат для создания нанопоровых чипов [37] и инструментарий, необходимый для их работы (проточная микросистема, микроэлектроника и т.п.) [38,39]. Нанопоровые чипы, разработанные ONT, содержат от 500 до 3000 белковых пор, встроенных в искусственные амфифильные мембраны, и позволяют проводить независимое измерение силы тока, протекающего через каждую нанопору. Нанопоровый чип интегрирован в одноразовую проточную ячейку (flow cell), которая является заменяемой частью нанопорового секвенатора. Первый коммерческий нанопоровый секвенатор MinION был предложен «ONT» в 2015 г. В настоящее время компания предлагает несколько нанопоровых секвенаторов разной производительности: MinION (производительность 10 – 30 млрд. нт на одну проточную ячейку в зависимости от используемого типа чипов), GridION (имеет до 5 проточных ячеек, производительность – до 150 млрд. нт за 48 ч) и PromethION (до 48 проточных ячеек, производительность – до 15 трлн. нт за 48 ч). MinION представляет портативный секвенатор (размер 10.5 × 2.3 × 3.3 см), который подключается к компьютеру через USB-разъем и рассчитан на одну проточную ячейку. Компания также объявила о разработке ещё более портативной версии нанопорового секвенатора - SmidgION, который рассчитан на работу с мобильными устройствами (в том числе таким, как смартфон). Уже в ранних работах молекулы РНК, наряду с молекулами ДНК, использовались для изучения прохождения нуклеиновых кислот через белковые нанопоры [23], что указывало на потенциальную возможность нанопорового секвенирования не только ДНК, но и РНК. Впервые возможность нанопорового секвенирования непосредствено РНК, без необходимости получения кДНК с помощью обратной транскрипции, была продемонстрирована компанией «ONT» в 2018 г. на примере polyA+РНК, выделенной из дрожжей [40]. Практически 80% «чтений», полученных в результате прямого секвенирования РНК, картировалось на геном дрожжей, а распределение длин «чтений» было одинаково с их распределением при секвенировании кДНК. В отличие от прямого секвенирования РНК с использованием SMRT-технологии, которое так и не получило развития, прямое нанопоровое секвенирование РНК уже применяется для секвенирования геномов РНК-вирусов [41] и транскриптомного анализа [42-44]. Нанопоровое секвенирование характеризуется достаточно длинными «чтениями». Так, при использовании одного из вариантов улучшенной технологии нанопорового секвенирования (известного как R9.0), медианное значение длины «чтения» составляет более 13 тыс. нт, максимальное – превышает 150 тыс. нт [45]. Основным недостатком нанопорового секвенирования является его невысокая точность. Несмотря на быстрый прогресс (в 2015 г. точность секвенирования составляла около 60% [10], а в 2018 – уже более 90% [42]), связанный с постоянным улучшением технологии нанопорового секвенирования и метода анализа первичных данных (например, использование бактериального мембранного белка CsgG для формирования нанопор и алгоритма Albacore для base-calling, основанного на использовании рекуррентной нейронной сети [45]), она существенно уступает точности SGS. Тем не менее, за счёт своей длины, «чтения» могут быть однозначно картированы на референсный геном с точностью, превышающей 97%, что позволяет уже сегодня успешно использовать нанопоровое секвенирование для детекции патогенных микроорганизмов и профилирования микробиомов [46-49]. 2. ИЗУЧЕНИЕ ТРАНСКРИПТОМА С ПОМОЩЬЮ ТЕХНОЛОГИЙ ОДНОМОЛЕКУЛЯРНОГО СЕКВЕНИРОВАНИЯ 2.1. Применение SMRT-секвенирования в транскриптомном анализе Исследование «ширины» транскриптома (многообразия транскриптов и их изоформ) с помощью SGS-технологий сталкивается со значительными трудностями, ассоциированными с проблемой точной реконструкции полноразмерных транскриптов из коротких «чтений» [16,50]. Так, сравнение 14 математических алгоритмов компьютеризованной «сборки» полноразмерных транскриптов из данных SGS, полученных для образцов личинок C. elegans и D. melanogaster и клеток линии HepG2, показало, что в первых двух случаях максимум 73% всех транскриптов могут быть реконструировано как полноразмерные (в среднем – 50 - 55%), а в последнем – максимум 61% (в среднем – 41%) [51]. Эти результаты также указывают на то, что количественные профили анализируемого транскриптома могут заметно различаться в зависимости от использованного алгоритма обработки первоначальных данных. Значительная длина «чтений» при SMRT-секвенировании (в среднем 10-20 тыс. нт) позволяет получать полноразмерные последовательности для подавляющего большинства транскриптов (например, у человека медианное значение длины транскрипта составляет около 2.5 тыс. нт [50], что делает возможным прямую детекцию их изоформ, образующихся в результате альтернативного сплайсинга, и обнаружение новых генов. Однако точность идентификации изоформ транскриптов и новых генов существенно ограничивалась высоким уровнем ошибки, характерным для SMRT-секвенирования. Для преодоления этого ограничения, Au и соавт. разработали математический алгоритм, позволяющий корректировать ошибки в длинных «чтениях», получаемых при SMRT-секвенировании транскриптома, с помощью коротких «чтений», получаемых при его секвенировании методами SGS [52]. Алгоритм, названный LSC, включает пять шагов: гомополимерную компрессию (замену последовательностей одного нуклеотида на единичный нуклеотид) длинных и коротких «чтений», контроль качества коротких «чтений», картирование коротких «чтений» на длинные, корректировку ошибок и декомпрессию последовательностей. Этот алгоритм был успешно реализован при исследовании изоформ транскриптов эмбриональных стволовых клеток человека: комбинирование и совместный анализ данных, полученных с использованием SMRT- и SGS-технологий позволили обнаружить 2103 изоформы, неаннотированных на тот момент в базе данных RefSeq или других базах данных, а также идентифицировать 273 новых гена [50]. Позднее был предложен более быстродействующий вариант LSC, названный LSCplus [53]. Для корректировки ошибок в длинных «чтениях» с помощью коротких «чтений» также были предложены алгоритмы: LoRDEC [54] (корректировка длинных «чтений» с помощью коротких, основанная на использовании графов де Брёйна); PacBioToCA, который являлся частью программного пакета Celera Assembler (в настоящее время больше не поддерживается); proovread (также основанный на картировании коротких «чтений» на длинные) [55]. Хотя точность SMRT-секвенирования существенно улучшилась за последние годы (в том числе из-за внедрения в практику секвенирования протокола Iso-Seq), комбинированный или, как его часто называют, гибридный подход, включающий совместное использование SMRT- и SGS-технологий, и сегодня широко используется в транскриптомном анализе (например, [56-62]). Его несомненным достоинством является отсутствие необходимости в референсном геноме (или в его полноте), недостатком – высокая стоимость из-за использования двух методов секвенирования. Протокол Iso-Seq, разработанный «Pacific Biosciences» в 2014 г., получил широкое распространение с момента его первого использования в 2015 г. [63] и является сегодня доминирующем подходом к анализу транскриптомов различного происхождения с использованием технологии SMRT-секвенирования [16]. Среди экспериментальных исследований, выполненных в 2018 г. с применением этого протокола, преобладают работы, посвященные альтернативному сплайсингу [56-60,64,65] и аннотации геномов [58,61,66-69]. Наряду с этим, SMRT-секвенирование по протоколу Iso-Seq также используется для изучения альтернативного полиаденилирования [70-72] и детекции химерных (или «слитых») генов (fusion genes) [73]. Использование SMRTbell-библиотек в протоколе Iso-Seq и последующая обработка получаемых CLR, результатом которой является набор RoI, представляющих консенсусные последовательности, существенно повышает конечную точность секвенирования. Так, SMRT-анализ по протоколу Iso-Seq транскриптома проростков сорго показал при сравнении с референсным геномом, что вероятность ошибки в расчёте на один нуклеотид составляет 2.34% и распределена следующим образом: однонуклеотидные замены – 0.64%, вставки – 1.07%, делеции – 0.63% [71]. Это позволяет при наличии референсного генома провести анализ «ширины» транскриптома SMRT-секвенированием без привлечения методов SGS. Алгоритм анализа, получивший название TAPIS (Transcriptome Analysis Pipeline for Isoform Sequencing), представляет собой итеративный процесс, который чередует картирование «чтений» на референсный геном и коррекцию ошибок [71]. Для картирования авторы использовали программу GMAP (Genome Mapping and Alignment Program) [74]. Анализ данных SMRT-секвенирования проростков сорго с использованием TAPIS позволил обнаружить более 11 тыс. новых сплайс-изоформ, сайты альтернативного полиаденилирования в приблизительно 11 тыс. экспрессирующихся генах и более 2100 новых генов [71]. Более того, существенное снижение ошибки SMRT-секвенирования при использовании протокола Iso-Seq позволило предложить алгоритм, получивший название ToFU (Transcript isOforms: Full-length and Unassembled), для анализа транскриптома de novo (то есть без использования референсного генома) без необходимости в коротких «чтениях» [75]. Алгоритм основан на анализе RoI и кластеризации их таким образом, чтобы получить набор консенсусных последовательностей, который и представляет набор транскриптов. Использование алгоритма ToFU для исследования транскриптома грибов вида Plicaturopsis crispa позволило идентифицировать почти 23 тыс. различных изоформ, представляющих около 9 тыс. транскрибируемых локусов. Сопоставление полученных последовательностей транскриптов с ранее аннотированным геномом Plicaturopsis crispa с помощью программы GMAP показало, что точность секвенирования составляет 0.22% (0.06% - замены, 0.04% - вставки, 0.12% - делеции) [75]. Следует однако отметить, что значительное число изоформ транскриптов, предсказываемых ToFU, может являться артефактами секвенирования, как показала их верификация методом количественной ПЦР при машинном обучении классификатора SQANTI (Structural and Quality Annotation of Novel Transcript Isoforms), предназначенного для отсеивания «артефактных транскриптов» при анализе данных SMRT-секвенирования [76]. В то время как SMRT-секвенирование per se или в сочетании с SGS широко используется для исследования альтернативного сплайсинга, альтернативного полиаденилирования, аннотации геномов и секвенирования транскриптомов de novo, оно практически не использовалось для количественного профилирования транскриптома. В работах, где транскриптомный анализ проводился с использованием гибридного секвенирования, сочетающего технологии SMRT и SGS, для количественного профилирования транскриптома использовались короткие «чтения», генерируемые SGS [62,77-81]. Одна из причин того, что SMRT-секвенирование не применяется для количественного профилирования, состоит в использовании протокола Iso-Seq, который предполагает фракционирование кДНК по размеру и последующую раздельную амплификацию фракций, что может существенно исказить оценку количества транскриптов по количеству RoI. Однако отказ от использования этого шага в протоколе Iso-Seq приведёт к предпочтительному секвенированию более коротких фрагментов [16]. Тем не менее, оценка дифференциальной экспрессии генов с помощью SMRT-секвенирования без использования шага фракционирования в протоколе Iso-Seq, по-видимому, в принципе возможна. При анализе транскриптомов вируса герпеса животных на разных стадиях литического цикла [82] и эндогенных вирусов человека при ВИЧ-инфекции [83], дифференциальную экспрессию генов вирусов оценивали используя количество RoI, полученных после обработки первоначальных данных SMRT-секвенирования. При этом фракционирование кДНК в протоколе Iso-Seq не проводилось [82,83]. Хотя это может завышать количество более коротких транскриптов, авторы, очевидно, исходили из предположения, что оно завышено в одинаковой пропорции в секвенируемых образцах. Насколько правомочен такой подход к дифференциальному профилированию транскриптомов, чей размер существенно превосходит размер транскриптома вирусов, в настоящее время не известно. Возможно, добавление в секвенируемые образцы «калибрантов» – молекул РНК с уникальной последовательностью и известной концентрацией (подобных тем, что разрабатываются в рамках проекта ERCC, «External RNA Controls Consortium» [84]) позволит проводить определение как относительного, так и абсолютного количество транскриптов по количеству RoI. При наличии линейки таких калибрантов, различающихся длиной молекул РНК, количественное профилирование транскриптомов с помощью SMRT-секвенирования может стать возможным как в абсолютных, так и относительных величинах даже в рамках протокола Iso-Seq. В любом случае, использование SMRT-секвенирования для количественного профилирования транскриптомов эукариот потребует достижения им производительности, не уступающей производительности SGS, чтобы обеспечить надёжную количественную детекцию низкокопийных транскриптов. 2.2. Анализ транскриптома с использованием нанопорового секвенирования Как и в случае с SMRT-секвенированием, использование нанопорового секвенирования в транскриптомном анализе было в первую очередь сфокусировано на идентификации изоформ транскриптов. Первое такое исследование было посвящено характеризации экспрессии генов со сложной экзон-интронной организацией, а именно генов Rdl, MRP, Mhc и Dscam1 дрозофилы [85], которые могут кодировать сотни и тысячи изоформ благодаря альтернативному сплайсингу. Авторы получали библиотеки полноразмерных кДНК транскриптов этих генов с помощью оптимизированной процедуры обратной транскрипции с последующей ПЦР со сменой матрицы (template-switching polymerase chain reaction, twPCR), которая обеспечивает избирательную амплификацию молекул кДНК, соответствующих полноразмерным транскриптам (full-length cDNA, FL-cDNA). Секвенирование FL-cDNA-библиотек на MinION и картирование полученных «чтений» на референсные последовательности с использованием алгоритма для выравнивания LAST [86] позволило обнаружить 7874 различных изоформ транскрипта гена Dscam1 (Dscam1 содержит 115 экзонов, 95 из которых могут участвовать в альтернативном сплайсинге и потенциально генерировать 30016 изоформ), а для генов Rdl, MRP и Mhc – 301, 337 и 112 изоформ, соответственно [85]. Аналогичный подход был также использован для анализа альтернативного сплайсинга гена BRCA1 человека с использованием мРНК из клеток лимфобластоидной линии [87]. FL-cDNA-библиотека была секвенирована на MinION и полученные «чтения» картированы на последовательность гена с помощью программы GMAP. В результате было обнаружено 32 изоформы транскрипта гена BRCA1 человека, из которых 20 изоформ не были описаны ранее. Нанопоровое секвенирование было также использовано для изучения транскриптома бакуловируса Autographa californica multiple nucleopolyhedrovirus, (AcMNPV) с использованием комбинации секвенирования кДНК-библиотеки и прямого секвенирования РНК [44]. Это позволило выявить 5 новых сплайс-изоформ известных транскриптов и 132 ранее не описанных транскрипта. Интересно, что секвенирование кДНК-библиотеки дало 324677 «чтений» (средняя длина 1053 нт), из которых 103133 были картированы на геном AcMNPV (используя программу GMAP; остальные «чтения» представляли транскрипты клетки-хозяина), в то время как прямое секвенирование РНК дало только 6482 «чтения» (средняя длина 614 нт; из них 2430 были картированы на вирусный геном) [44]. В ноябре 2018 г. была опубликована работа Seki и соавт. [42], в которой был впервые выполнен анализ транскриптома клеток человека (клеточная линия аденокарциномы лёгких LC2/ad) с использованием нанопорового секвенирования и идентификации изоформ транскриптов. В результате секвенирования FL-cDNA-библиотеки было получено 532956 «чтений», которые были картированы на транскрипты в базе данных RefSeq с использованием двух алгоритмов для выравнивания – LAST и BWA [88]. Как отметили авторы работы, использование LAST более предпочтительно, так как он позволил картировать большую долю «чтений» на референсный транскриптом (38.3% vs. 33.9%) с покрытием транскриптов «чтениями» 0.8 и более. В результате было идентифицировано 6018 изоформ транскриптов, из которых 158 – ранее не аннотированных. Кроме того, проведённый анализ позволил выявить 151 химерный ген [42]. Аналогично SMRT-секвенированию, нанопоровое секвенирование используется в гибридном секвенировании как технология, «комплементарная» SGS [89-92]. Применительно к анализу транскриптома, корректировка длинных «чтений» с помощью коротких (с использованием программы proovread) позволила снизить ошибку нанопорового секвенирования с 12% до 0 - 2% [93]. Нанопоровое секвенирование также было применено для секвенирования транскриптома de novo [94], для чего авторами был разработан алгоритм анализа длинных «чтений», основанный на их кластеризации на основе схожести без обращения к референсному геному или транскриптому, получивший название CARNAC-LR (Clustering coefficient-based Acquisition of RNA Communities in Long Reads). Кластеризация длинных «чтений», полученных в результате нанопорового секвенирования РНК из мозга мышей (всего 1256967 «чтений») и сравнение полученных результатов с результатами картирования этих «чтений» на референсный геном (с помощью программы BLAT [95]) показало приблизительно 80% совпадение, что указывает на сложность сборки больших транскриптомов de novo из данных нанопорового секвенирования без корректировки ошибок секвенирования с помощью референсного генома или транскриптома. Вероятно, это связано с существенной долей однонуклеотидных замен, вставок и делеций в «чтениях», которые, в отличие от SMRT-секвенирования, не могут быть корректированы при получении консенсусной последовательности из многих «субчтений». В случае нанопорового секвенирования, максимально возможное количество «субчтений» в настоящее время не превышает двух (2D- или 1D2-секвенирование). В то время как SMRT-технология характеризуется избирательностью, заключающейся в предпочтительном секвенировании более коротких последовательностей ДНК (и, соответственно, необходимостью использования протокола Iso-Seq для того, чтобы уменьшить эту избирательность), нанопоровое секвенирование, по-видимому, подобной избирательности не имеет. В недавней работе Oikonomopoulos и соавт. [96], нанопоровое секвенирование FL-cDNA-библиотек, полученных для смесей 92 полиаденилированных транскриптов (отобранных в рамках проекта ERCC [84]) с заданной концентрацией, показало, что количество «чтений» хорошо согласуется с ожидаемой концентрацией соответствующего транскрипта (коэффициент корреляции Пирсона rp равнялся 0.98) и не зависит от его длины или GC-состава молекул кДНК. Интересно, что в этом случае использование алгоритма для выравнивания LAST также давало наилучшее соответствие между количеством «чтений» и ожидаемым количеством транскрипта в сравнении с такими алгоритмами, как Margin-Align [97], BWA, BLASR [98], BLAST [99] и Smith-Waterman [100]. Отсутствие избирательности нанопорового секвенирования по длине и GC-составу транскриптов делает принципиально возможным оценку их относительного и/или абсолютного количества. В качестве меры количества каждого транскрипта используют число «чтений», картируемых на его последовательность [42,43], что требует, соответственно, наличия референсного транскриптома или генома. Подобный подход был реализован при исследовании диффференциальной экспрессии генов дрожжей Saccharomyces cerevisiae [43]. Используя прямое нанопоровое секвенирование РНК, было получено ~530 и ~620 тыс. «чтений» с медианной длиной 1150 и 1263 нт, соответственно, для полиаденилированной РНК дрожжевых клеток, культивируемых в различных условиях (при избытке глюкозы или в присутствии этанола). «Чтения» были картированы на геном S. cerevisiae с помощью алгоритма выравнивания GraphMAP, что позволило идентифицировать 5433 различных транскриптов (91% от их ожидаемого числа). Алгоритм GraphMAP [101] был специально разработан для картирования на референсный геном длинных «чтений», получаемых при нанопоровом секвенировании и характеризующихся относительно высокой ошибкой секвенирования (в данном случае она составляла 12% [43]). Отношение количества «чтений», приходящихся на каждый транскрипт при различных условиях культивирования, было использовано для анализа дифференциальной экспрессии генов [43]. В другой публикации [42] нанопоровое секвенирование FL-cDNA-библиотеки, полученной для полиаденилированной РНК культивируемых клеток аденокарциномы легкого человека, было использовано для количественного профилирования их транскриптома. В качестве метрики авторы использовали RPM (Reads Per Million; доля «чтений», картированных на данный транскрипт, от их общего количества, умноженная на миллион). Количественный профиль транскриптома, полученный с использованием нанопорового секвенирования, показал хорошее соответствие с профилем, полученным секвенированием с помощью SGS (rp = 0.91) [42]. При этом для SGS в качестве метрики использовали TPM (Transcripts Per Million mapped reads). TPM, наряду с RPKM и FPKM, часто используется для количественной оценки результатов SGS и рассматривается как метрика, устраняющая сдвиг в оценке количества транскриптов, возможный при использовании RPKM- и FPKM-метрик [102]. Следует отметить, что валидация количественного профилирования транскриптома культивируемых клеток аденокарциномы человека методом ОТ-ПЦР (обратная транскрипция и ПЦР в реальном времени) на выборке из 44 транскриптов показала хорошее соответствие с результатами нанопорововго секвенирования (rp = 0.82) [42]. Таким образом, нанопорове секвенирование позволяет проводить количественное профилирование транскриптомов эукариот, включая клетки человека. Очевидно, что добавление в секвенируемые образцы «РНК-калибрантов» сделает возможным наряду с оценкой относительного количества транскриптов также и определение их абсолютного количества. ЗАКЛЮЧЕНИЕ Одномолекулярное секвенирование с использованием технологий SMRT- и нанопорового секвенирования существенно расширяет возможности исследования многообразия изоформ транскриптов, образующихся в результате альтернативного сплайсинга и/или полиаденилирования, секвенирования транскриптомов de novo, аннотации геномов и идентификации химерных генов, особенно при сочетании с SGS. Применительно к количественному профилированию транскриптома, SMRT-технология имеет очевидные ограничения, связанные с предпочтительным секвенированием коротких фрагментов кДНК и с неопределённостями в оценке количества транскриптов, вносимыми необходимостью фракционирования кДНК по размеру в рамках протокола Iso-Seq. Напротив, нанопоровое секвенирование не имеет таких ограничений и может быть использовано для количественного профилирования транкриптома, особенно в формате прямого секвенирования РНК, в котором нанопоровый секвенатор может выступать в качестве молекулярного счётчика, позволяющего идентифицировать транскрипт с высокой селективностью и оценить его относительную представленность в транскриптоме. При использовании РНК-калибрантов – молекул РНК с уникальными последовательностями и известной концентрацией – нанопоровое секвенирование позволит определять абсолютное количество транскрипта каждого вида в анализируемых образцах. БЛАГОДАРНОСТИ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013–2020 годы. ЛИТЕРАТУРА
|