|
Алгоритм определения последовательности белка с использованием комбинации методов деградации по Эдману и панорамного масс-спектрометрического анализа Научно-исследовательский институт биомедицинской химии имени В. Н. Ореховича, 119121, Москва, Погодинская ул. 10 ;*e-mail: vladlen@ibmh.msk.su Ключевые слова: деградация по Эдману; панорамный масс-спектрометрический анализ; de novo секвенирование DOI: 10.18097/BMCRM00087 ВВЕДЕНИЕ
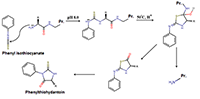
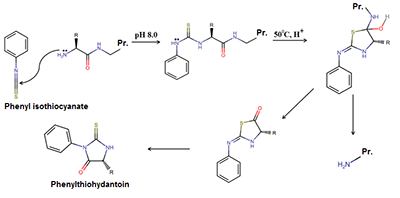
Несмотря на стремительный рост числа расшифрованных геномов, задача расшифровки белковой последовательности (или секвенирование) непосредственно по имеющемуся белку не потеряла своей актуальности. Это особенно важно при определении аминокислотной последовательности вариабельных фрагментов антител, а также белков организмов с неизвестным геномом, последовательности различных биологически активных пептидов и т.д. Один из старейших методов, используемых для этого – деградация по Эдману [1]. В ходе секвенирования проводят пошаговое (по одной аминокислоте) химическое отщепление меченого присоединением фенилизотиоцианата (ФИТЦ) N-концевого аминокислотного остатка (рис. 1) и идентифицируют его при помощи хроматографии. Основными проблемами в этом методе являются невысокая скорость анализа и нарастающая вероятность ошибок при проведении большого числа итераций, так как, несмотря на высокий выход реакции, не все N-концевые остатки модифицируются, и постепенно достоверность определения отщеплённого остатка падает. Другим способом расшифровки первичной структуры белков и пептидов является масс-спектрометрический анализ пептидов, полученных при расщеплении белка с последующей идентификацией пептидов масс-спектров процедурой de novo секвенирования. Существует достаточное число алгоритмов и программ, позволяющих провести подобный анализ [2]. Однако, по понятным причинам результаты работы таких программ очень сильно зависят от качества спектров и часто не дают однозначного результата. Кроме того, при учете всех возможных масс-спектрометрических артефактов время счёта критически возрастает, а точность идентификации существенно падает. В настоящей работе рассмотрен алгоритм, позволяющий объединить преимущества таких методов как деградация по Эдману и масс-спектрометрическое de novo секвенирование.
МАТЕРИАЛЫ И МЕТОДЫ В качестве тестового белка в работе использовали белок-реагент для кожного теста "Диаскинтест" (DSTP) [3], применяемый в диагностике туберкулеза. Это рекомбинантный белок размером 209 аминокислотных остатков. Экспериментальную часть выполняли по следующей схеме: белок предварительно подвергали расщеплению трипсином (метод описан ранее [4]), затем последовательно проводили 5 шагов деградации по Эдману. Исходную смесь пептидов и смесь, полученную после каждого из шагов деградации по Эдману, подвергали протеомному панорамному анализу. Пробу пептидов, полученных при ферментативном гидролизе белка трипсином, предварительно выпаривали в вакуумном концентраторе Concentrator Plus («Eppendorf», Германия). Реакцию присоединения ФИТЦ к N-концевой α-аминогруппе пептида проводили в буфере, содержащем пиридин:ФИТЦ:ddH2O, pH 8.0 в соотношении 2.5:0.5:1. Реакцию проводили при температуре 50°C в течение 10 мин. По окончании реакции проводили отмывку от побочных продуктов реакции раствором, содержащим гексан:этилацетат в соотношении 10:1. Для проведения реакции кислотного гидролиза по первой пептидной связи использовали трифторуксусную кислоту, реакцию проводили при температуре 50ºС в течение 3 мин. По окончании реакции отбирали аликвоту для LC-MS/MS анализа. На этом этапе цикл начинали сначала. Перед аналитическим разделением пептиды наносили на обогащающую колонку Accalaim Precolumn (размер частиц 0.5 мм × 3 мм, 5 мк) («Thermo Scientific», США) при скорости потока 10 мкл/мин в течение 5 мин в изократическом режиме подвижной фазы Б (2% ацетонитрил, 0.1% муравьиная кислота). Протеомный панорамный анализ пептидов осуществляли с использованием хроматографической ВЭЖХ системы Ultimate 3000 RSLCnano («Thermo Scientific»). Пептиды разделяли на ВЭЖХ колонке Acclaim Pepmap® C18 (размер частиц 75 мкм × 150 мм, 2 мкм) («Thermo Scientific») в градиентном режиме элюирования. Градиент формировали подвижной фазой А (0.1% муравьиная кислота) и подвижной фазой Б: (80% ацетонитрил, 0.1% водный раствор муравьиной кислоты) при скорости потока 0.3 мкл/мин. Колонку промывали 2% подвижной фазой Б в течение 10 мин, после чего линейно увеличивали концентрацию подвижной фазы Б до 37% за 45 мин, затем линейно увеличивали концентрацию фазы Б до 90%, после этого аналитическую колонку промывали для уравновешивания 2% фазой Б в течение 10 мин. Общая длительность анализа составляла 65 мин. Масс-спектрометрический анализ проводили на гибридном орбитальном масс-спектрометре Orbitrap Q-exactive («Thermo Scientific») в режиме положительной ионизации в источнике NESI («Thermo Scientific»). Напряжение на эмиттере 2.1 кВ. Панорамное сканирование проводили в диапазоне масс от 400 m/z до 2000 m/z, тандемное сканирование фрагментных ионов от нижней границы 110 m/z до верхней границы, определяемой зарядным состоянием прекурсорного иона. Для тандемного сканирования учитывали только ионы от z = 2+ до z = 6+ по зарядному состоянию. Максимальное число разрешённых для изоляции ионов в режиме MS2 было установлено не более 20. Максимальное время накопления для прекурсорных ионов составило 50 мс, для фрагментных ионов – 110 мс. Полученные после панорамного сканирования MS/MS спектры анализировали как собственным программным обеспечением, описанным ниже, так и двумя программами de novo секвенирования Novor [5] и PepNovo+ [6]. Для всех программ использовали сходный набор параметров: точность определения массы первичного иона 5 ppm, точность определения массы фрагментов 0.02 Да; в качестве возможных модификаций рассматривали карбамидметилирование цистеина, дезаминирование глутамина и аспарагина, а также окисление метионина. Программы обрабатывали все 6 проб одновременно. Для анализа покрытия использовали программу ProteoCat [7]. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Первым этапом работы была попытка использовать уже существующие программы de novo секвенирования. На рисунке 2 представлено покрытие последовательности белка, полученное в результате работы программ Novor и PepNovo+. Общий процент покрытия составил 24% для программы Novor и 39% для PepNovo+. Однако, в отличие от PepNovo+, программа Novor всегда пытается дать единственное, но полноценное решение, тогда как PepNovo+ может выдавать достоверную часть последовательности (TAG) и остаточные массы (с N и/или C концов). Если рассмотреть только фрагменты пептидов, предсказанных программой Novor, для которых поаминокислотный счёт больше 50 при минимальной длине фрагмента в 6 остатков, степень покрытия возрастает до 43%. Тем не менее, даже при таком допущении часть возможных пептидов, наличие которых ожидаемо после трипсинолиза, не идентифицируется процедурой de novo секвенирования.
Если более подробно рассмотреть предсказанные пептиды, то иногда видны следы того, что деградация по Эдману проводилась. Наиболее яркий пример, полученный программой PepNovo+, представлен в таблице 1 на пептиде QKQELDELSTNIR (напомним, что пару I/L программы de novo секвенирования не различают). Видно, что N-концевой фрагмент QKQE легко восстанавливается. Однако, та же таблица демонстрирует и имеющуюся проблему. Пептиды 4, 5, 6 и 14 заведомо не могут пройти барьер в 5 ppm, определённый как точность измерения массы первичного иона. Пептид 11 также не пройдёт этот барьер, но данный случай, скорее всего, действительно связан либо с ошибкой измерения, либо с особенностями работы программы PepNovo+, в то время как остальные, вероятнее всего, обусловлены наличием масс-спектрометрических артефактов, либо неучтённых модификаций. Например, в исследуемой выборке можно предположить наличие следующих модификаций помимо 4-х, указанных выше: формилирование, цитрулирование аргинина, аддукт формальдегида, ацетилирование. Иногда модификации накладываются друг на друга, и предположить, что именно вызывает изменение массы пептида, затруднительно.
Последующий алгоритм определения аминокислотной последовательности белка включает следующие процедуры. Для сравнения спектров между собой и объединения их в кластеры для каждого из спектров вычисляют сигнатуру из пиков, которые могут соответствовать Y-ионам (масса пика равна сумме масс комбинации из 20 аминокислотных остатков с учётом возможных модификаций плюс 19.0184). Затем рассматривают все последовательности таких пиков таким образом, чтобы расстояние между ними соответствовало массе одного остатка, либо их комбинации (в текущем варианте комбинация не могла иметь массы более 300), при условии, что отклонения укладываются в заданную точность (в данной работе 0.02 Да). В качестве сигнатуры отбирают последовательность пиков с минимальным RMSD от «идеального» и максимальной длины. Сравнение сигнатур идёт от пика наименьшей массы в сторону увеличения. Пики считают совпадающими, если их массы различаются в пределах установленной точности измерения. При этом допускается пропуск 1-2 пиков, но не больше чем на дельту масс в 300 Да. Совпадающими считают сигнатуры, у которых число совпавших пиков более установленного порога (в данной работе 7). Сигнатура может быть представлена в виде аминокислотной последовательности, но, если промежуток между пиками соответствует больше чем одному аминокислотному остатку, критериев выбора их очередности в программе нет. Данная процедура позволяет отнести к конкретному кластеру значительно большее число спектров, чем полноценная процедура de novo секвенирования. Так, если с помощью программы PepNovo+ из 4083 имеющихся вторичных спектров для кластера пептида QKQELDELSTNIR можно отобрать 14 спектров, то число отобранных пептидов описанной процедурой будет значительно больше (265). Среди них и пептид с массой, соответствующей последовательности QKQELDELSTNIR (моноизотопная масса 1573.818 Да, 23 спектра).
Для каждого выявленного кластера значения масс первичных ионов сортируют и удаляют повторы, после чего, рассчитав разницу масс, можно восстановить последовательность аминокислотных остатков с N-конца. Принцип отбора продемонстрирован в таблице 2. Например, если начинать с наименьшей массы, то, перемещаясь от нижней левой ячейки по диагонали матрицы разности масс, доходим до ячейки с дельтой масс, соответствующей конкретному аминокислотному остатку (выделено красным цветом, значение точности при этом зависит от точности измерения для первичного иона, в данной работе 5 ppm). Если такого значения не находится, то переходим к следующему (большему по массе, но не больше, чем на массу минимального по массе аминокислотного остатка) пептиду как к стартовому и повторяем процедуру. Если аминокислотный остаток идентифицируется, то следующую итерацию начинаем с пептида, по которому идентифицировали предыдущий остаток (выделен серым цветом). Для надёжности проводим обратную процедуру – от большей массы к меньшей. При наличии разночтений результат необходимо анализировать. Обычно это связано с тем, что в кластере присутствуют как модифицированные (например, окисленный метионин) по N-концевым аминокислотным остаткам пептиды, так и немодифицированные. В данной работе при восстановлении N-концевого фрагмента не использовали массы, соответствующие сумме двух и более остатков, но это также можно использовать. С-концевую сигнатуру также пересчитывают в фрагмент последовательности и вычисляют промежуточную масса (N-TAG – промежуточная масса – С-TAG). Если промежуточная масса соответствует конкретному аминокислотному остатку, то её также можно считать идентифицированной. Результат представлен на рисунке 3. В настоящей работе некоторые из пептидов (рис. 3) имели пересечение между N- и С-концевыми фрагментами. Кроме того, некоторые небольшие пептиды были полностью идентифицированы только за счёт Y-ионов. Также видно, что идентифицированные фрагменты пептидов 9 и 11 не триптические. Вероятно, это связано с наличием побочной неспецифичной активности у фермента [8]. Следует помнить, что задачи достоверной и доказательной идентификации C-концевого фрагмента или полного пептида перед нашей программой не стояло. В первую очередь она призвана выявить последовательность N-концевого фрагмента с помощью метода деградации по Эдману. Но даже несмотря на то, что процедура идентификации C-концевого фрагмента может быть существенно улучшена, покрытие DSTP увеличивается до 57.5% (рис. 2) без учёта фрагмента 179-206, для которого вычислена масса, но сам он не идентифицирован.
ЗАКЛЮЧЕНИЕ С помощью предлагаемого алгоритма метод деградации по Эдману можно с успехом применять для разрешения последовательности неизвестного белка или идентификации смеси пептидов, особенно, если комбинировать его с другими методами de novo секвенирования. Из приведённых данных видно, что неразрешёнными остаются очень короткие и очень протяжённые фрагменты белковой цепи. Если первый вариант имеет смысл решать с использованием протеазы со специфичностью отличной от трипсина, то второй – путём увеличения числа шагов деградации по Эдману, так как пептид при этом будет укорачиваться, и вероятность его детекции возрастёт. В отличие от классического секвенирования методом деградации по Эдману, то, что по мере увеличения числа шагов увеличивается число непрореагировавшего белка, и идентифицировать уходящий аминокислотный остаток становится затруднительно, в данном варианте не принципиально. Кроме того, на каждом последующем шаге количество всё более и более редуцированного пептида будет уменьшаться, но при достаточно большом количестве исходного белка это также не должно быть проблемой, пока оставшаяся длина пептида достаточна для идентификации C-концевого фрагмента. БЛАГОДАРНОСТИ Работа выполнена в рамках Программы фундаментальных научных исследований государственных академий наук на 2013-2020 годы. Масс-спектрометрические данные были получены в ИБМХ на оборудовании ЦКП “Протеом человека”, поддержанного Минобрнауки России в рамках выполнения соглашения №14.621.21.0017 (уникальный идентификатор работ RFMEFI62117X0017). ЛИТЕРАТУРА
|