Algorithm of Protein Sequence Determination by Combination of the Edman Degradation Method and Shotgun Mass Spectrometry
Institute of Biomedical Chemistry, 10 Pogodinskaya str., Moscow, 119121 Russia; *e-mail: vladlen@ibmh.msk.su
Keywords: Edman degradation; shotgun mass spectrometry; de novo sequencing
DOI:10.18097/BMCRM00087
An algorithm combining advantages of the Edman degradation method and de novo mass-spectrometric sequencing was developed. The protein from the “Diaskintest” diagnostic test was used for analysis. The protein was digested with trypsin and 5 steps of Edman degradation were carried out sequentially for the mixture of peptides. At each stage, the resulting mixture was analyzed by shotgun mass spectrometry analysis. The results of mass-spectrometry were analyzed both by the well-known de novo sequencing programs Novor and PepNovo+, and by own program that clustered individual spectra with a C-terminal signature formed by Y-ions. This approach allows us to determine confidently the amino acid sequence of the N-terminal part of the peptides obtained after the protein hydrolysis by trypsin.
|
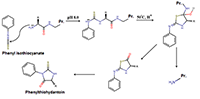
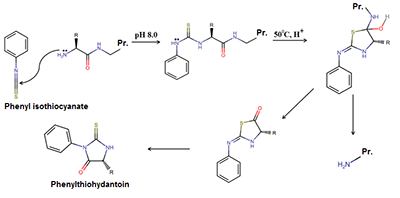
Figure 1.
The scheme of the N-terminal amino acid residue cleavage reaction (Edman degradation).
|


|
Figure 2.
The coverage of the DSTP sequence by peptides identified by de novo sequencing programs Novor and PepNovo+ (PN+). Nov.2 –only the part of the peptide predicted by Novor, with single amino acid accuracy > 50 (minimum length is 6 residues) was used for coverage. In the case of PepNovo+ the symbol “X” marked the matching residual mass with a correctly identified TAG. EDMAN: the N-terminal fragment calculated by the mass difference of peptides is marked red; green designates the C-terminal fragment; “X” designates a fragment for which the mass is known, but sequence not determined.
|


|
Figure 3.
Identification of DSP tryptic peptides by the Edman degradation method. Black color indicates the peptide obtained after trypsinolysis. Red color indicates the N-terminal fragment, calculated from the difference of mass of peptides. Green color indicates the C-terminal fragment (or peptide completely) calculated from the signature, built on probable Y-ions. <AL> - the order of amino acid residues is unknown. |
|
CLOSE

|
Table 1.
The identification of the QKQELDELSTNIR peptide and its fragments by the PepNovo+. The peptide fragments were obtained by Edman degradation. Red color highlighted amino acid residues determined by mass difference of peptides. (the QKQE sequence determined for the N-terminus of the peptide). The numbers designate the residual mass minus the mass of known amino acid residues up to this position.
|
|
CLOSE
|
Table 2.
The analysis of peptides combined by the C-terminal signature in the QKQELDELSTNIR cluster. Red color highlighted difference of masses that allow to determine amino acid residues. Gray background indicates masses of peptides that differ by one amino acid residue (the QKQE sequence determined for N-terminus of the peptide).
|
ACKNOWLEDGEMENTS
This work was performed within the framework of the Program for Basic Research of State Academies of Sciences for 2013–2020. Mass spectrometric measurements were performed using the equipment of the“Human Proteome” Core Facility at the Institute of Biomedical Chemistry supported by the Ministry of Education and Science of the Russian Federation (agreement 14.621.21.0017, Project ID RFMEFI62117X0017).
REFERENCES
- Edman, P. (1950). Method for determination of the amino acid sequence in peptides. Acta chem. scand, 4(7), 283-293. DOI
- Vyatkina, K. V. (2018). De novo секвенирование белков и пептидов: алгоритмы, приложения, перспективы. Biomedical Chemistry: Research and Methods, 1(1), e00005. DOI
- Kiselev, V. I., Baranovskiĭ, P. M., Rudykh, I. V., Shuster, A. M., Mart'ianov, V. A., Mednikov, B. L., ... & Slogotskaia, L. V. (2009). Clinical trials of the new skin test Diaskintest for the diagnosis of tuberculosis. Problemy tuberkuleza i boleznei legkikh, (2), 11-16.
- Mikurova A.V., Novikova S.E., Skvortsov V.S., Alekseychuk N.N., Rybina A.V., Miroshnichenko Yu.V. (2017) The sequence coverage in different methods of mass spectrometry data analysis obtained on model proteins, Biomeditsinskaya khimiya, 63(5), 397-404. DOI
- Ma, B. (2015). Novor: real-time peptide de novo sequencing software. Journal of the American Society for Mass Spectrometry, 26(11), 1885-1894. DOI
- Frank, A., & Pevzner, P. (2005). PepNovo: de novo peptide sequencing via probabilistic network modeling. Analytical chemistry, 77(4), 964-973. DOI
- Skvortsov, V. S., Alekseychuk, N. N., Miroshnichenko, Y. V., & Rybina, A. V. (2018). pIPredict Version 2: New Features and PTM Analysis. Biomedical Chemistry: Research and Methods, 1(2), e00009. DOI
- Keil, B. (2012). Specificity of proteolysis. Springer Science & Business Media.