|
СОДЕРЖАНИЕ |
pIPredict версия 2: новые возможности и работа с PTM
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича,
119121 Москва, ул. Погодинская, 10 стр. 8, *e-mail: vladlen@ibmh.msk.su
Ключевые слова: белок; пептид; изоэлектрическая точка; посттрансляционные модификации; предсказание свойств DOI: 10.18097/BMCRM00009 ВВЕДЕНИЕ
Программа pIPredict [1] была создана как инструмент предсказания изоэлектрической точки пептидов и белков, она также может быть использована для генерации простейшей карты виртуального 2D электрофореза. Метод предсказания pI основан на использовании уравнения Хендерсона-Хассельбаха [2]. Несмотря на то, что его использование даёт хорошие результаты, само уравнение является аппроксимацией, а многие факторы, влияющие на значение pI (например, зависимости от температуры и ионной силы раствора), традиционно игнорируются. Как правило, различные варианты отличаются только шкалой констант диссоциации (pKa) соответствующих групп аминокислотных остатков, «рассчитанных» различными способами. Это может быть титрование модельных пептидов [3], квантово-химические расчёты или подбор значений pKa на большом массиве данных об экспериментальных значениях pI для известных белков и/или пептидов [1,4]. Применяют также и другие методы (не связанные с уравнением Хендерсона-Хассельбаха), например, метод опорных векторов (SVM, Support Vector Machines) [5] или искусственные нейронные сети (ANN; Artificial Neural Networks) [1]. Однако, если последовательность сильно отличается по размеру от тех, что использовали при обучении, SVM и ANN не дают удовлетворительного результата. Так как ранее встроенный в программу pIPredict метод с использованием ANN был обучен на небольших пептидах и плохо работает на белках, от него было решено отказаться. В основном, новые и обновлённые функции программы pIPredict связаны с работой с пострансляционными модификациями (PTM). Кроме того, в программу также добавлена возможность коррекции электрофоретического сдвига по молекулярному весу по методу, описанному нами ранее [6].
ОПИСАНИЕ ПРОГРАММЫ Программа pIPredict 2.0 написана на языке JAVA, распространяется в виде исполняемого jar-архива и доступна по адресу http://www.ibmc.msk.ru/LPCIT/pIPredict.
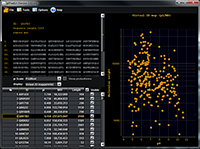
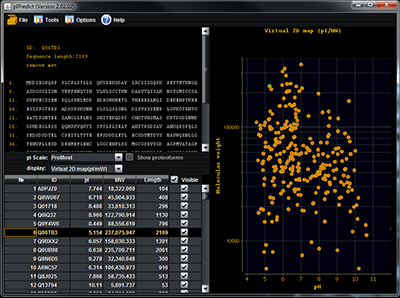
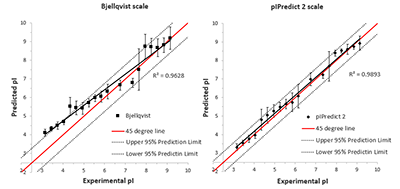
Программа имеет развитой графический интерфейс пользователя (рис. 1) и следующий набор возможностей: 1. Модуль загрузки последовательностей белков и пептидов из файлов в формате FASTA или простом текстовом формате с указанием модификаций аминокислотных остатков, список которых доступен в руководстве пользователя. Формат позволяет определить произвольное число PTM, описание которых может редактироваться пользователем. 2. Модуль предсказания pI с использованием нескольких шкал значений pKa. В настоящей версии к имеющимся ранее шкалам (Bjellqvist [7] и ещё 7 шкал) добавлены: новая версия шкалы pIPredict (рис. 2), рассчитанная на основе скорректированной обучающей выборки pIPredict версии 1.0, и шкала программы ProMoST [8], позволяющая предсказывать pI с учётом фосфорилирования аминокислотных остатков.
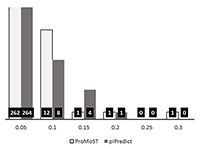
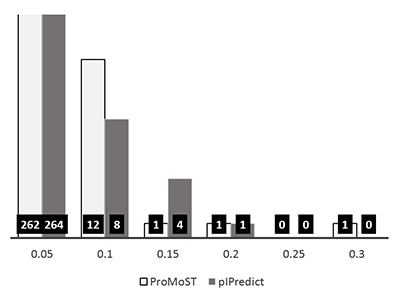
Шкалы pIPredict и ProMoST имеют значения pKa для учёта модификаций, вносящих дополнительную диссоциирующую группы (например, фосфорилирование), в остальных шкалах могут учитываться только модификации, элиминирующие такие группы, либо модифицирующие одни аминокислотные остатки в другие (например, дезаминирование аспарагина). При предсказании pI имеется возможность учитывать облигатные химические модификации (например, карбамидметилирование цистеина, что важно при оценке pI пептидов в масс-спектрометрических экспериментах) и удаление N-концевого остатка метионина (важно для оценки pI белков при анализе данных электрофореза). Как и в случае фосфорилирования удаление N-концевого метионина даёт эффект только при использовании шкал pIPredict и ProMoST. Как правило, этими изменениями можно пренебречь, так как изменения превышают среднюю ошибку предсказания примерно в 5% случаях (рис. 3); тем не менее это может быть важно для каждого конкретного белка. Имеется возможность загрузки собственной шкалы pKa (или модификации любой из имеющихся шкал).
3. Модуль расчёта зависимости усреднённого заряда белков (пептидов) от pH для выбранных белков и модуль расчёта значений усреднённого заряда при заданном pH для каждой аминокислотной последовательности из загруженного набора белков. 4. Модуль коррекции электрофоретического сдвига по молекулярной массе [6]. 5. Модуль вариативного предсказания pI для набора модификаций, полученных из мастерной аминокислотной последовательности белка по заданным правилам (рис. 4). Данная функция может быть использована как для предсказания положения модифицированной формы на карте виртуального 2D электрофореза, так и для формирования гипотезы, какая конкретно форма белка может наблюдаться в эксперименте.
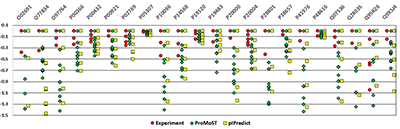
Например, (рис. 4) при анализе белков тканей почки Bos Taurus [9] для ряда белков были обнаружены несколько вариантов положения идентифицированных белков (выделены красным). Если поварьировать 4 возможных изменения (фосфорилирование остатков треонина или серина и спонтанное дезаминирование остатков глутамина или аспарагина), сделать предсказания для pI шкалами ProMoST (зелёный) и pIPredict (жёлтый) и сравнить изменения относительно варианта с крайним значением pI в основной области (в данном случае делается предположение, что данный вариант соответствует мастерной форме белка), видно, что в ряде случаев (O02691, O77834, P10096, P14568, P41976, Q28035, Q95KZ6) можно предсказать, какой форме будет соответствовать обнаруженная экспериментально протеоформа. Правда, следует отметить, что в настоящее время у авторов нет неопровержимых доказательств в пользу того, что это не случайные совпадения. Однако, по мере развития программы возможности такого рода предсказания будут исследоваться отдельно. Важным для данной функции являются точность предсказания pI (для шкалы pIPredict априорная средняя точность предсказания 0.1 значения величины pH), а также способ, которым проводится калибровка данных электрофореза. Часто полученные данные калибруются по предсказанным значениям pI для группы идентифицированных белков. Все данные, полученные в результате работы программы pIPredict версии 2.0 могут быть экспортированы в виде текстовых файлов с разделителем, что позволяет легко использовать их в любой программе анализа электронных таблиц. БЛАГОДАРНОСТИ Работа выполнена при финансовой поддержке Программы фундаментальных научных исследований государственных академий наук на 2013–2020 годы. ЛИТЕРАТУРА
|