Идентификация белков плазмы крови человека с использованием внутренних пептидных стандартов в панорамном протеомном анализе
Научно-исследовательский институт биомедицинской химии имени В.Н. Ореховича, 119121, Москва, Погодинская ул. 10; *e-mail: victor.zgoda@gmail.com
Ключевые слова: масс-спектрометрия; идентификация белков; панорамный протеомный анализ; внутренний петидный стандарт
DOI:10.18097/BMCRM00093
Использование метода панорамной жидкостной хроматографии-масс-спектрометрии (ЖХ-МС/МС) позволяет идентифицировать в сложных протеомах до нескольких тысяч белков. Однако значительная часть протеома остается недоступной для идентификации из-за отсутствия или плохого качества МС/МС спектров. Описываемый в данной работе метод повышает вероятность идентификации белков плазмы крови человека путем выравнивания хроматографических данных образца смеси протеолитических пептидов и этого же образца, разбавленного синтетическими пептидами. Такая идентификация происходит в результате сопоставления тандемных масс-спектров синтетических пептидов с соответствующими выровненными хроматографическими пиками протеолитических пептидов. Используя данный подход, мы выявили 19 низко представленных белков плазмы крови человека, соответствующих 19 синтетическим пептидам, выбранным для исследования. Проверка идентификаций методом мониторинга диссоциативных переходов с изотопно-мечеными стандартами подтвердила наличие в плазме 17 белков.
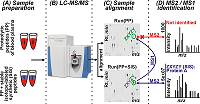
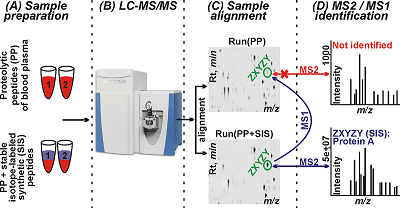
|
Рисунок 1.
Схема эксперимента по идентификации белков плазмы крови человека путем выравнивания хроматографических данных образцов смесей протеолитических пептидов и с этими же образцами, разбавленными синтетическими пептидами. Основные этапы: А) пробоподготовка; Б) проведение панорамной жидкостной хроматографии-масс-спектрометрии; В) выравнивание хроматограмм образцов в программе «Progenesis LC-MS»; Г) поиск идентификаций белков в программе «Mascot» и назначение тандемных масс-спектров (МС2) синтетических пептидов соответствующим хроматографическим пикам протеолитических пептидов.
|

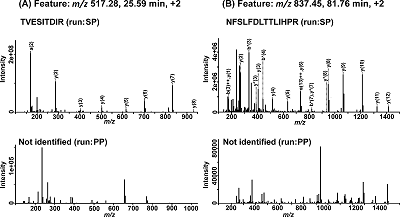
|
Рисунок 2.
Идентифицированные и неидентифицированные тандемные масс-спектры, соответствующие одному участку выровненных относительно друг друга хроматограмм образцов смесей протеолитических пептидов (SamplePP) и этих же образцов, содержащих синтетические пептиды (SamplesSP). A) Идентифицированный (для SamplesSP) МС2 спектр пептида TVESITDIR и неидентифицированный (для SamplePP) МС2 спектр, относящиеся к участку выровненных хроматограмм с m/z 517.28. (B) Идентифицированный (для SamplesSP) МС2 спектр пептида NFSLFDLTTLIHPR и неидентифицированный (для SamplePP) МС2 спектр, относящиеся к участку выровненных хроматограмм с m/z 837.45.
|


|
Рисунок 3.
Идентификация в плазме крови человека фактора свертывания крови X (P00742) методом мониторинга диссоциативных переходов. Показаны ионные хроматограммы нативного и изотопно-меченого синтетического (heavy) пептида ETYDFDIAVLR из программы Skyline.
|
|
ЗАКРЫТЬ

|
Таблица 1.
Список белков/пептидов идентифицированных в плазме крови.
|
ФИНАНСИРОВАНИЕ РАБОТЫ
Работа выполнена в рамках Программы президиума РАН «Протеомно-метаболомный профиль здорового человека».
ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ
К данной статье приложены дополнительные материалы, свободно доступные в электронной версии (http://dx.doi.org/10.18097/BMCRM00093) на сайте журнала.
ЛИТЕРАТУРА
- Domon, B., & Aebersold, R. (2006). Mass spectrometry and protein analysis. Science, 312(5771), 212-217. DOI
- James, P., Quadroni, M., Carafoli, E., & Gonnet, G. (1993). Protein identification by mass profile fingerprinting. Biochemical and biophysical research communications, 195(1), 58-64. DOI
- Pappin, D. J., Hojrup, P., & Bleasby, A. J. (1993). Rapid identification of proteins by peptide-mass fingerprinting. Current biology, 3(6), 327-332. DOI
- Henzel, W. J., Watanabe, C., & Stults, J. T. (2003). Protein identification: the origins of peptide mass fingerprinting. Journal of the American Society for Mass Spectrometry, 14(9), 931-942. DOI
- Camerini, S., & Mauri, P. (2015). The role of protein and peptide separation before mass spectrometry analysis in clinical proteomics. Journal of chromatography A, 1381, 1-12. DOI
- Miroshnichenko, Y. V., Petushkova, N. A., Teryaeva, N. B., Lisitsa, A. V., Zgoda, V. G., Belyaev, A. Y., & Potapov, A. A. (2015). Identifi cation of Central Nervous System Proteins in Human Blood Serum and Plasma. Bulletin of experimental biology and medicine, 160(1), 35-39. DOI
- Michalski, A., Damoc, E., Hauschild, J. P., Lange, O., Wieghaus, A., Makarov, A., ... & Horning, S. (2011). Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Molecular & Cellular Proteomics, 10(9), M111-011015. DOI
- Miroshnichenko, Y. V., Petushkova, N. A., Moskaleva, N. E., Teryaeva, N. B., Zgoda, V. G., Ilgisonis, E. V., & Belyaev, A. Y. (2015). The possibility of using the PlasmaDeepDive™ MRM-panel in clinical diagnostics. Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 9(3), 283-289. DOI
- Michalski, A., Cox, J., & Mann, M. (2011). More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC− MS/MS. Journal of proteome research, 10(4), 1785-1793. DOI
- Laskay, U. A., Lobas, A. A., Srzentić, K., Gorshkov, M. V., & Tsybin, Y. O. (2013). Proteome digestion specificity analysis for rational design of extended bottom-up and middle-down proteomics experiments. Journal of proteome research, 12(12), 5558-5569. DOI
- Geiger, T., Cox, J., & Mann, M. (2010). Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Molecular & Cellular Proteomics, 9(10), 2252-2261. DOI
- Sajic, T., Liu, Y., & Aebersold, R. (2015). Using data‐independent, high‐resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. PROTEOMICS–Clinical Applications, 9(3-4), 307-321. DOI
- Gillet, L. C., Navarro, P., Tate, S., Röst, H., Selevsek, N., Reiter, L., ... & Aebersold, R. (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & Cellular Proteomics, 11(6), O111-016717. DOI
- Schubert, O. T., Gillet, L. C., Collins, B. C., Navarro, P., Rosenberger, G., Wolski, W. E., ... & Aebersold, R. (2015). Building high-quality assay libraries for targeted analysis of SWATH MS data. Nature protocols, 10(3), 426. DOI
- Conrads, T. P., Anderson, G. A., Veenstra, T. D., Paša-Tolić, L., & Smith, R. D. (2000). Utility of accurate mass tags for proteome-wide protein identification. Analytical chemistry, 72(14), 3349-3354. DOI
- Palmblad, M., Ramström, M., Markides, K. E., Håkansson, P., & Bergquist, J. (2002). Prediction of chromatographic retention and protein identification in liquid chromatography/mass spectrometry. Analytical chemistry, 74(22), 5826-5830. DOI
- Stanley, J. R., Adkins, J. N., Slysz, G. W., Monroe, M. E., Purvine, S. O., Karpievitch, Y. V., ... & Dabney, A. R. (2011). A statistical method for assessing peptide identification confidence in accurate mass and time tag proteomics. Analytical chemistry, 83(16), 6135-6140. DOI
- Pridatchenko, M. L., Tarasova, I. A., Guryca, V., Kononikhin, A. S., Adams, C., Tolmachev, D. A., ... & Zubarev, R. A. (2009). Use of models of biomacromolecule separation in AMT database generation for shotgun proteomics. Biochemistry (Moscow), 74(11), 1195. DOI
- Bączek, T., & Kaliszan, R. (2009). Predictions of peptides' retention times in reversed‐phase liquid chromatography as a new supportive tool to improve protein identification in proteomics. Proteomics, 9(4), 835-847. DOI
- Kunda, P. B., Benavente, F., Catalá-Clariana, S., Giménez, E., Barbosa, J., & Sanz-Nebot, V. (2012). Identification of bioactive peptides in a functional yogurt by micro liquid chromatography time-of-flight mass spectrometry assisted by retention time prediction. Journal of Chromatography A, 1229, 121-128. DOI
- Moruz, L., Staes, A., Foster, J. M., Hatzou, M., Timmerman, E., Martens, L., & Käll, L. (2012). Chromatographic retention time prediction for posttranslationally modified peptides. Proteomics, 12(8), 1151-1159. DOI
- Lobas, A. A., Verenchikov, A. N., Goloborodko, A. A., Levitsky, L. I., & Gorshkov, M. V. (2013). Combination of Edman degradation of peptides with liquid chromatography/mass spectrometry workflow for peptide identification in bottom‐up proteomics. Rapid Communications in Mass Spectrometry, 27(3), 391-400. DOI
- Goloborodko, A. A., Mayerhofer, C., Zubarev, A. R., Tarasova, I. A., Gorshkov, A. V., Zubarev, R. A., & Gorshkov, M. V. (2010). Empirical approach to false discovery rate estimation in shotgun proteomics. Rapid Communications in Mass Spectrometry: An International Journal Devoted to the Rapid Dissemination of Up‐to‐the‐Minute Research in Mass Spectrometry, 24(4), 454-462. DOI
- Bateman, N. W., Goulding, S. P., Shulman, N. J., Gadok, A. K., Szumlinski, K. K., MacCoss, M. J., & Wu, C. C. (2014). Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Molecular & Cellular Proteomics, 13(1), 329-338. DOI
- Ting, Y. S., Egertson, J. D., Payne, S. H., Kim, S., MacLean, B., Käll, L., ... & MacCoss, M. J. (2015). Peptide-centric proteome analysis: an alternative strategy for the analysis of tandem mass spectrometry data. Molecular & Cellular Proteomics, 14(9), 2301-2307. DOI
- Hood, C. A., Fuentes, G., Patel, H., Page, K., Menakuru, M., & Park, J. H. (2008). Fast conventional Fmoc solid‐phase peptide synthesis with HCTU. Journal of peptide science: an official publication of the European Peptide Society, 14(1), 97-101. DOI
- MacLean, B., Tomazela, D. M., Shulman, N., Chambers, M., Finney, G. L., Frewen, B., ... & MacCoss, M. J. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics, 26(7), 966-968. DOI
- Vizcaíno, J. A., Csordas, A., Del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., ... & Xu, Q. W. (2015). 2016 update of the PRIDE database and its related tools. Nucleic acids research, 44(D1), D447-D456. DOI
- Lange, V., Picotti, P., Domon, B., & Aebersold, R. (2008). Selected reaction monitoring for quantitative proteomics: a tutorial. Molecular systems biology, 4(1), 222. DOI
- Gianazza, E., Tremoli, E., & Banfi, C. (2014). The selected reaction monitoring/multiple reaction monitoring-based mass spectrometry approach for the accurate quantitation of proteins: clinical applications in the cardiovascular diseases. Expert review of proteomics, 11(6), 771-788. DOI
- Carr, S. A., Abbatiello, S. E., Ackermann, B. L., Borchers, C., Domon, B., Deutsch, E. W., ... & Liebler, D. C. (2014). Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Molecular & Cellular Proteomics, 13(3), 907-917. DOI