|
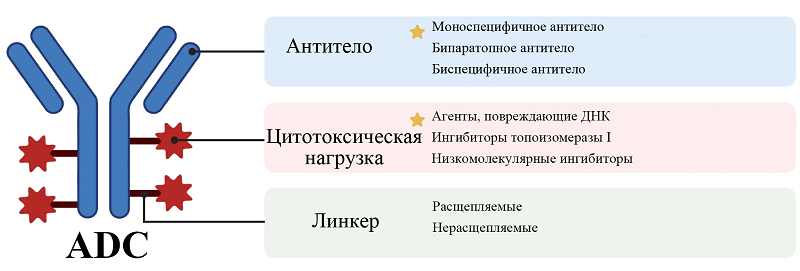
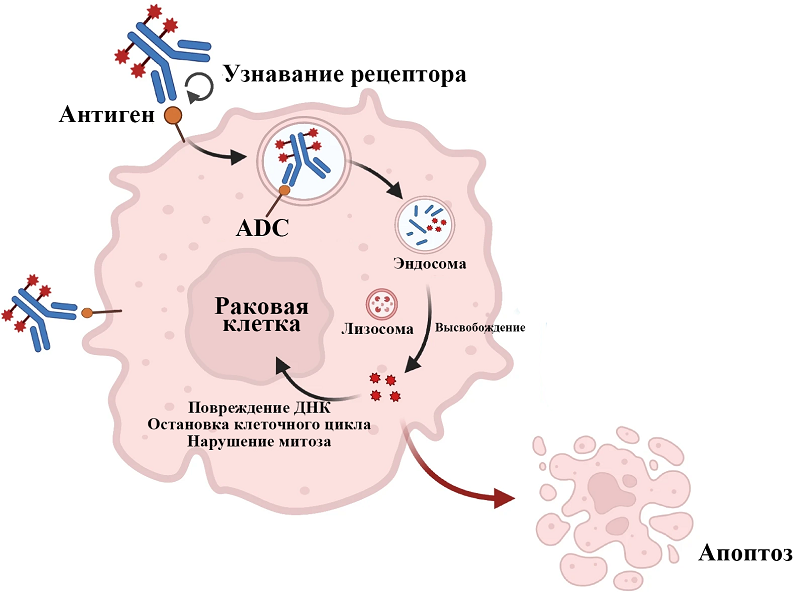
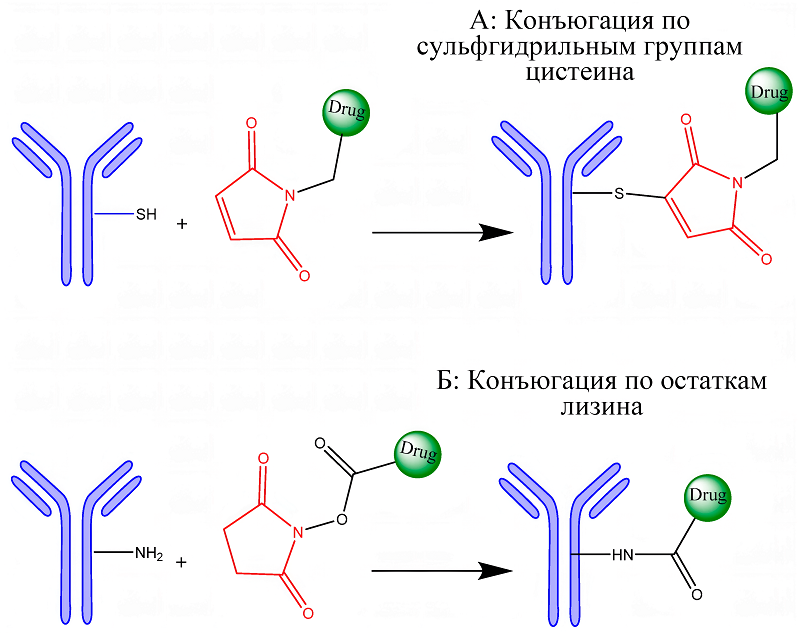
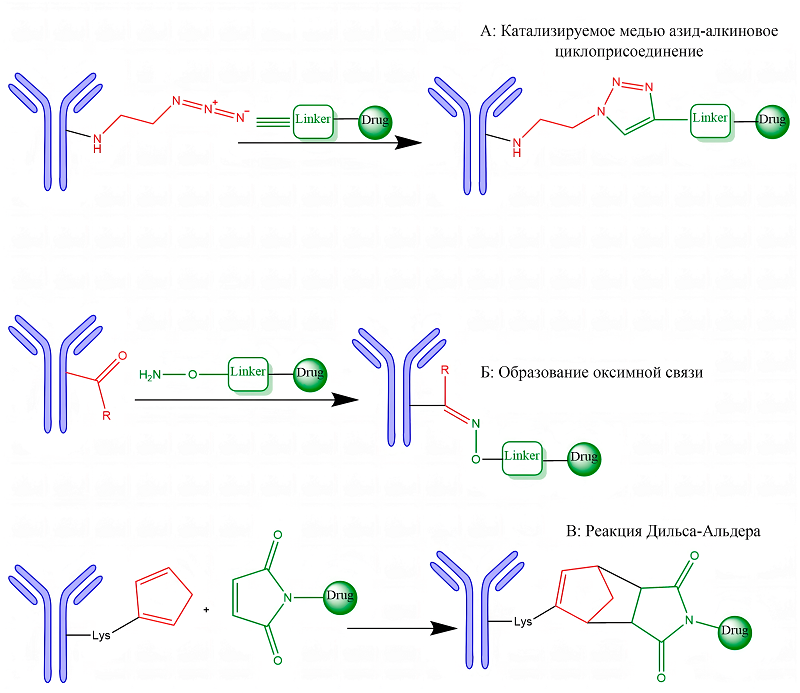
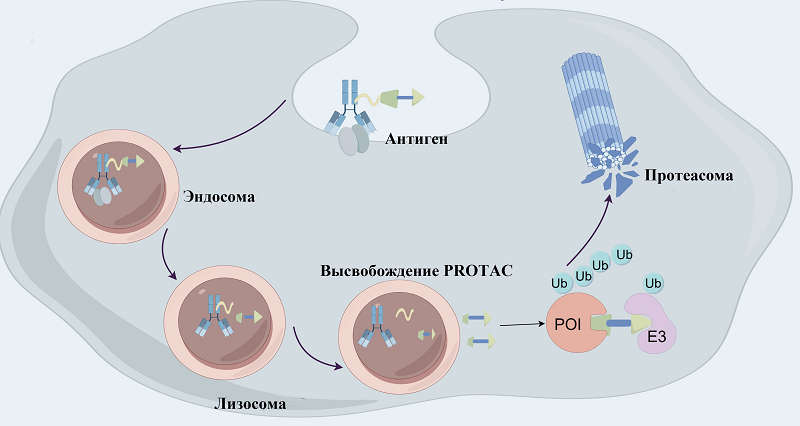
СОДЕРЖАНИЕ 1. КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО (ADC) 2. БИСПЕЦИФИЧЕСКИЕ АНТИТЕЛА: ПОВЫШЕНИЕ СЕЛЕКТИВНОСТИ ЗА СЧЕТ МУЛЬТИВАЛЕНТНОГО ТАРГЕТИНГА 3. МАЛЫЕ КАРКАСНЫЕ БЕЛКИ КАК АЛЬТЕРНАТИВНАЯ ПЛАТФОРМА ДЛЯ ТАРГЕТНОЙ ТЕРАПИИ СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Рисунок 5Две основные стратегии конъюгации в синтезе ADC. (A) Конъюгация с цистеином: восстановление межцепочечных дисульфидных связей приводит к образованию свободных тиоловых групп, которые реагируют с функционализированными малеимидом лекарственными линкерами с образованием стабильных тиоэфирных связей. (Б) Конъюгация с лизином: ε-аминогруппы лизинов, экспонированных в растворителе, реагируют с активированными эфирами (например, NHS-эфирами) лекарственных линкеров с образованием стабильных амидных связей. [105] (CC BY-SA 4.0). Рисунок 6Схематическая иллюстрация стратегий сайт-специфической конъюгации для ADC, включая (A) катализируемое медью циклоприсоединение азида к алкину, (Б) образование оксимной связи и (В) циклоприсоединение Дильса-Альдера, обеспечивающее селективное и стабильное связывание лекарственного средства с антителом [105] (CC BY-SA 4.0). Рисунок 7. Механизм действия конъюгатов DAC [129] (CC BY-SA 4.0). Таблица 1Одобренные ADC-препараты на мировом рынке Таблица 2Сравнительная характеристика и эволюция линкерных систем в ADC Таблица 3Одобренные ADC-препараты на основе биспецифических антител на мировом рынке |
НОВЫЕ ПОКОЛЕНИЯ АНТИТЕЛ И КАРКАСНЫХ БЕЛКОВ: СОЗДАНИЕ ВЫСОКОСЕЛЕКТИВНЫХ КОНЪЮГАТОВ ДЛЯ ОНКОЛОГИИ Институт биологического приборостроения, Пущинский научный центр биологических исследований, 142290, Московская обл., Пущино, ул. Институтская, 7; *e-mail: loskutova-i@mail.ru Ключевые слова: конъюгаты антитело-лекарство (ADC); таргетная терапия; биспецифические антитела; малые каркасные белки; линкеры, цитотоксические агенты; сайт-специфическая конъюгация; PROTAC DOI: 10.18097/BMCRM00305 ВВЕДЕНИЕ Таргетная терапия злокачественных новообразований представляет собой одно из наиболее динамично развивающихся направлений современной онкологии. За последние десятилетия подходы к созданию терапевтических конъюгатов претерпели значительную эволюцию – от первых поколений препаратов с ограниченной эффективностью к современным высокоспецифичным конструкциям нового поколения. К первому поколению терапевтических конъюгатов относятся препараты на основе традиционных моноклональных антител, которые характеризовались рядом фундаментальных ограничений. Во-первых, это использование цитотоксических агентов со значениями IC50 (концентрация, вызывающая 50% ингибирования) ~ 1–10 нМ, таких как калихеамицин [1]. В контексте ADC такая активность считалась недостаточной, поскольку для уничтожения клетки требовалось доставить большое количество молекул конъюгата, что повышало их общую токсичность [1]. Во-вторых, применение химически лабильных линкеров, в частности гидразоновых, которые были чувствительны не только к кислому pH лизосом, но и к гидролизу в плазме крови [2]. Эта недостаточная селективность активации являлась фундаментальным недостатком. В-третьих, ключевым ограничением стала гетерогенность и размер молекул первого поколения. Значительный размер полноразмерных антител (~150 кДа) затруднял их транспорт через физиологические барьеры опухоли и достижение раковых клеток в ее глубине, при этом использовавшиеся методы неспецифической конъюгации (например, по остаткам лизина) создавали гетерогенную смесь молекул с разным числом цитотоксических агентов [3]. Эта гетерогенность делала поведение препарата in vivo непредсказуемым. Преодоление этих барьеров стало возможным с появлением платформ нового поколения, целенаправленно разработанных для устранения перечисленных ограничений. К отличительным признакам новых поколений относятся: переход к би- и мультиспецифическим таргетным модулям для повышения селективности, использование малых каркасных белков (аффибоди, DARPins, аднектины) для улучшенного проникновения в опухоль и сайт-специфической конъюгации, внедрение стабильных, контролируемо расщепляемых линкерных систем, расширение арсенала полезных нагрузок, а также применение сайт-специфических методов конъюгации для получения гомогенных препаратов [4]. Существенный прорыв был достигнут в разработке новых форматов целевых носителей – би- и триспецифических антител, нацеленных на несколько мишеней. Несмотря на сохранение крупных размеров, эти конструкции повышают избирательность действия благодаря активации терапевтического эффекта при одновременном связывании с несколькими разными опухолевыми антигенами. Параллельно с усовершенствованием антительных платформ активно развивается направление, связанное с созданием альтернативных каркасных белков [5]. Эти молекулы характеризуются компактными размерами, обычно находящимися в диапазоне от 1 кДа до 20 кДа. Благодаря малой молекулярной массе они демонстрируют превосходную способность к глубокому проникновению в опухолевую ткань и более быструю кинетику распределения по сравнению с традиционными антителами. Кроме того, их монодоменная структура и высокая стабильность позволяют осуществлять сайт-специфическую конъюгацию с цитотоксическими агентами, обеспечивая получение гомогенных препаратов с воспроизводимыми фармакологическими свойствами. Важным преимуществом является возможность их эффективного производства в бактериальных системах экспрессии, что значительно снижает стоимость производства по сравнению с полноразмерными антителами, требующими использования сложных эукариотических систем. Значительный прогресс в разработке конъюгатов достигнут за счет усовершенствования всех структурных компонентов. Созданы линкерные системы с многоуровневой регуляцией расщепления, сочетающие pH-зависимую активацию с ферментативным расщеплением под действием катепсина B или β-глюкуронидазы, что обеспечивает селективность действия в опухолевом микроокружении [6]. Параллельно расширен арсенал цитотоксических агентов, включая высокоактивные ингибиторы микротрубочек нового поколения (тазаметостат), индукторы иммуногенной клеточной гибели (митоксантрон) и блокаторы транскрипционных процессов (α-аманитин). Совокупность этих усовершенствований позволила создать препараты с оптимизированным терапевтическим индексом, где высокая цитотоксичность сочетается с избирательностью доставки в опухолевую ткань. Особого внимания заслуживают принципиально новые технологические платформы, расширяющие саму парадигму таргетной терапии. Среди них PROTAC-ADC для направленной деградации белков, олигонуклеотидные конъюгаты для подавления экспрессии генов и фотоиммуноконъюгаты, активируемые светом [7]. Эти подходы позволяют не просто доставлять цитотоксические агенты, а тонко управлять внутриклеточными процессами. Настоящий обзор систематизирует современные данные о разработке высокоселективных конъюгатов для онкологии, с особым акцентом на стратегиях повышения их эффективности и безопасности. В работе детально проанализированы ключевые компоненты конъюгатов, механизмы действия перспективных конструкций, рассмотрены актуальные проблемы дизайна и пути их решения, а также представлена критическая оценка новейших технологических платформ и сохраняющихся вызовов в этой быстро развивающейся области. 1. КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО (ADC) Конъюгаты антител с лекарственными средствами (ADC) являются одним из быстроразвивающихся видов онкологических препаратов (рис. 1), представляя собой высокоэффективное лекарство и идеальную комбинацию химио- и иммунотерапии. С момента клинического применения первого ADC гемтузумаба озогамицина в 2000 году этот класс препаратов быстро развивался и оказался очень многообещающим [8]. Благодаря своим преимуществам направленной доставки некоторых очень мощных химиотерапевтических агентов он продемонстрировал не только значительную терапевтическую активность при тяжело поддающихся лечению рефрактерных заболеваниях, но также мощную активность с пролонгацией при раннем лечении заболевания.
Согласно данным на 2025 год, во всем мире одобрено для клинического применения более 20 ADC (табл. 1). Точное число варьирует в зависимости от страны и регуляторного органа (FDA, EMA, PMDA и др.), а также от того, включаются ли дополнительные показания для одного и того же препарата.
1.1. Механизм действия Конъюгаты ADC (антитело-лекарственное средство) состоят из антитела, цитотоксической нагрузки и химического линкера [25]. Как только ADC достигает клеток-мишеней, моноклональное антитело (mAb) распознает антигены клеточной поверхности и связывается с ними. Комплекс ADC-антиген затем интернализуется внутрь раковой клетки путем эндоцитоза с образованием ранней эндосомы, которая после созревания образует поздние эндосомы и, наконец, сливается с лизосомами. Цитотоксическое лекарственное средство затем высвобождается из конъюгата с моноклональным антителом посредством химической реакции или ферментативного расщепления в лизосомах и оказывает цитотоксический эффект, вызывая апоптоз или гибель клеток (рис. 2) [26].
В дополнение к цитотоксическим свойствам ADC, Fc-часть моноклонального антитела способствует развитию иммунозависимой цитотоксичности, такой как антитело-зависимая клеточно-опосредованная цитотоксичность (ADCC), антитело-зависимый фагоцитоз (ADP) и комплемент-зависимая цитотоксичность (CDC) [27]. Технологии генной инженерии позволяют в настоящее время усиливать эффекторную функцию антитела в Fc-области. Кроме того, связывание компонента антитела ADC со специфическим антигенным эпитопом раковых клеток может ингибировать последующую сигнальную трансдукцию антигенного рецептора [28]. Например, антитело трастузумаб связывается с рецептором HER2 раковых клеток и ингибирует образование гетеродимера между HER2 и HER1, HER3 или HER4, блокируя пути сигнальной трансдукции выживания и пролиферации клеток, что вызывает апоптоз клеток [29]. 1.2. Целевой антиген Выбор соответствующего целевого антигена является обязательным условием разработки ADC. Во-первых, антиген должен экспрессироваться преимущественно в опухолевых клетках, чтобы снизить нецелевую токсичность [30]. Во-вторых, связывание с целевым антигеном должно приводить к интернализации комплекса антиген-антитело. Кроме того, в идеале он должен находиться на поверхности клетки, чтобы его можно было распознать, и, наконец, он не должен быть секреторным, поскольку секретируемый антиген в кровообращении может вызвать связывание ADC вне опухолевых участков [31, 35]. Целевыми антигенами ADC, лицензированными FDA, являются CD19, CD22, CD30, CD33 и CD79b при гематологических злокачественных новообразованиях, а также HER2, TROP-2, нектин-4, тканевой фактор и рецептор фолиевой кислоты альфа (FRα) при солидных опухолях. При этом «идеальный» фрагмент антитела также должен способствовать эффективной интернализации, иметь высокое сродство к антигену, длительный период полувыведения из плазмы и проявлять низкую иммуногенность [33]. mAb имеют большой размер и составляют более 90% массы любого ADC. При таком соотношении наблюдается снижение распределения или проникновения препарата в здоровые ткани, включая те, которые обычно функционируют как метаболизирующие органы и органы выведения [34]. Однако в случае опухоли такой проблемы не возникает, поскольку неполноценный эндотелий опухолевых сосудов обладает высокой проницаемостью [35]. Важным аспектом является поиск антигенов, экспрессируемых в стромальной части опухоли или на неопластических клетках, но не на стволовых клетках здоровых тканей. Это позволяет снизить токсичность для быстро обновляющихся здоровых тканей (костный мозг, слизистые). Например, мишень B7-H3 (CD276) широко экспрессируется в строме различных солидных опухолей и на некоторых опухолях нейроэктодермального происхождения, но минимально присутствует на нормальных тканях, что делает ее перспективной мишенью для ADC нового поколения.
1.3. Выбор линкерной последовательности как важная часть оптимизации препарата На разнообразные свойства ADC, включая специфичность, стабильность, эффективность и токсичность, влияют также свойства линкера [36]. Используемые на сегодняшний день типы линкеров представлены в таблице 2. Расщепляемые линкеры либо химически лабильны (гидразоновая группа и дисульфидный «мостик»), либо ферментативно лабильны. Гидролиз гидразоновой связи в линкере происходит преимущественно в лизосомах и эндосомах при интернализации, с периодическим гидролизом в плазме, что приводит к нецелевой системной токсичности [37]. Дисульфидный тип линкера может быть стабильным в плазме, специфически высвобождая активный «полезный груз» в раковых клетках с повышенным уровнем восстановленного глутатиона [37]. Чувствительные к ферментам линкеры имеют сродство к лизосомальной протеазе, которая обычно сверхэкспрессируется в раковых клетках, что обеспечивает точное высвобождение лекарственного средства в клетках после интернализации [38]. ADC с нерасщепляемыми линкерами устойчивы к химическому или ферментативному расщеплению в плазме и требуют полной деградации антитела в поздних эндосомах и лизосомах для высвобождения полезной нагрузки. Следовательно, ADC с нерасщепляемыми линкерами могут иметь наименьшую нецелевую системную токсичность из-за повышенной стабильности в плазме, становясь наиболее подходящими для лечения опухолей с гомогенной экспрессией антигена [39]. Некоторые из ADC были разработаны так, чтобы оказывать желаемый «нецелевой эффект» для «убийства сторонних наблюдателей», распространяя цитотоксический эффект на клетки с низкой или отрицательной экспрессией антигена в непосредственной близости от опухоли. Для работы этого механизма решающее значение имеют несколько характеристик молекул ADC, а именно: расщепляемый линкер и неполярная, свободно проникающая через мембрану полезная нагрузка. Например, трастузумаб дерукстекан (T-DXd) имеет расщепляемый ферментативный линкер, в отличие от трастузумаба эмтанзина, который имеет тиоэфирный нерасщепляемый линкер. Полезные нагрузки, такие как дерукстекан, MMAE или майтансиноид DM4, проникают сквозь клеточные мембраны и могут диффундировать из клеток после расщепления в лизосомах. Следовательно, в силу свойств своего компонента T-DXd обладает «эффектом свидетеля» и, таким образом, эффективен даже при опухолях с низкой экспрессией HER2, тогда как трастузумаб эмтанзин, благодаря своим свойствам, имеет более низкий профиль токсичности [40]. Напротив, чтобы уменьшить нежелательную системную токсичность молекул полезной нагрузки, проникающих из опухолевых клеток, можно использовать ионизируемые полезные нагрузки (например, содержащие карбоновые кислоты). На данный момент трастузумаб эмтанзин является единственным одобренным ADC, в котором используется нерасщепляемый линкер, тогда как остальные ADC оснащены расщепляемыми линкерами. Сейчас активно идет модификация и разработка новых линкерных связей.
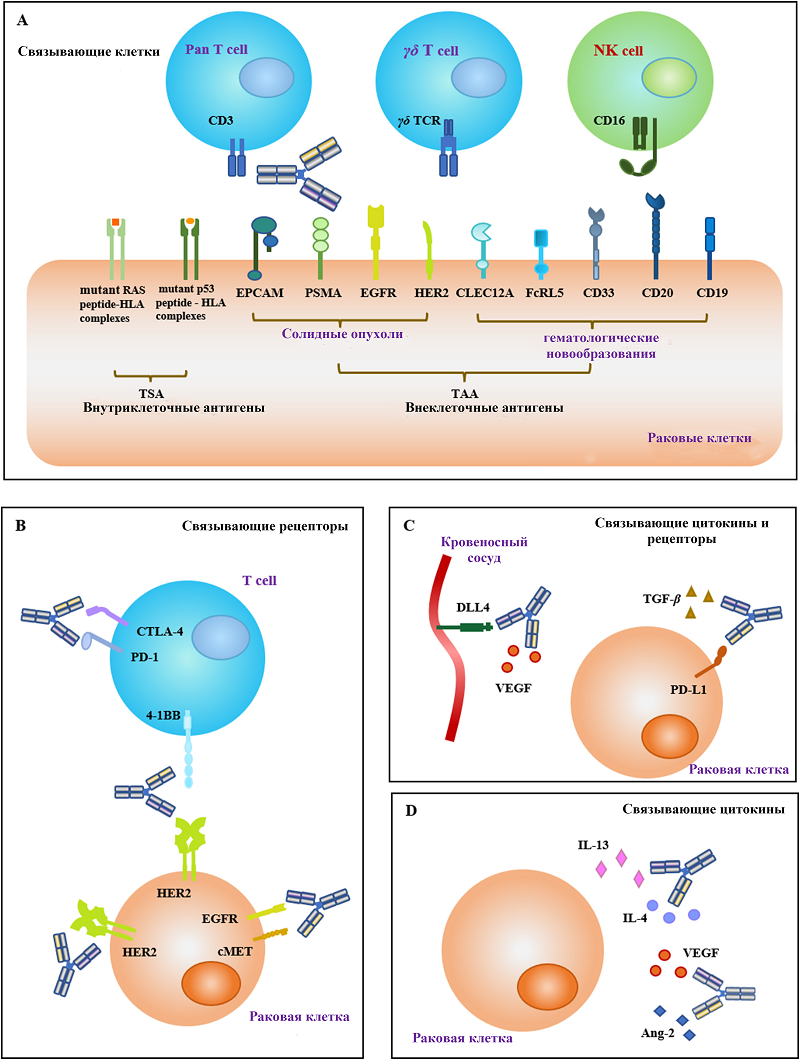
Современные разработки сосредоточены на создании «умных» линкеров, обеспечивающих двойной или тройной контроль высвобождения. Такие линкеры требуют одновременного выполнения нескольких условий для активации (например, низкое значение pH и наличие сайта для расщепления соответствующей протеазой), что радикально повышает их стабильность в плазме и снижает системную токсичность [41]. Кроме того, активно развиваются линкеры, чувствительные к внешним воздействиям, таким как ультрафиолетовое излучение или ультразвук, для управления высвобождением лекарства в заданной локации [42]. 1.4. Цитотоксические соединения Цитотоксические соединения в идеале должны обладать следующими свойствами: высокая эффективность, высокая цитотоксическая активность in vitro (ингибирование 50% от максимального), высокая стабильность в системе кровообращения, достаточная растворимость в водной среде и биохимические свойства, наличие реакционноспособных групп, обеспечивающих конъюгацию с антителом, низкая иммуногенность, небольшая молекулярная масса и длительный период полураспада [43]. В одобренных в настоящее время ADC в качестве т.н. «полезной нагрузки» используют в основном два класса цитотоксических препаратов: ингибиторы микротрубочек и агенты, повреждающие ДНК. Ауристатин и майтанзин, используемые в качестве полезной нагрузки, являются цитотоксическими агентами, которые действуют как ингибиторы тубулина. Ауристатин — синтетический аналог доластатина. Существует два основных производных ауристатина: монометил ауристатин E (MMAE) и монометил ауристатин F (MMAF). Ключевое структурное различие между ними заключается в С-концевой аминокислоте: MMAF содержит заряженный остаток фенилаланина, в то время как MMAE — нейтральный. Это различие напрямую влияет на их биофизические свойства: наличие заряда у MMAF препятствует его пассивной диффузии через клеточные мембраны, локализуя его действие внутри клетки, тогда как нейтральный MMAE способен покидать клетку и проникать в соседние, реализуя так называемый "эффект свидетеля" [27]. Эти два продукта структурно различаются: фенилаланин, присутствующий на С-конце, не позволяет MMAF проникать сквозь мембраны, тогда как MMAE может выходить из клетки и, таким образом, диффундировать в соседние клетки и убивать их [27]. К ADC с полезной нагрузкой MMAE относятся брентуксимаб ведотин, полатузумаб ведотин, энфортумаб ведотин и тисотумаб ведотин (табл. 1). Белантамаб мафодотин использует MMAF в качестве цитотоксической полезной нагрузки (белантамаб в настоящее время снят с продажи). Майтансиноиды — природные цитотоксические агенты, выделенные из коры Maytenus serrata, имеющей макролидную структуру. Майтансиноидные полезные нагрузки, DM1 и DM4, являются компонентами трастузумаба эмтанзина и мирветуксимаба соравтансина соответственно (табл. 1) [44]. Калихеамицины, пирролобензодиазепины и ингибиторы топоизомеразы являются агентами, повреждающими ДНК, которые действуют путем индуцирования двуцепочечных разрывов ДНК, способствуя образование сшивок ДНК [45]. И гемтузумаб озогамицин, и инотузумаб озогамицин содержат в качестве полезной нагрузки N-ацетил-γ-калихеамицин. Калихеамицины принадлежат к классу мощных противоопухолевых агентов, которые расщепляют ДНК сайт-специфическим двухцепочечным способом. Пирролобензодиазепины представляют собой еще один класс антибиотиков, полученных из видов Streptomyces. Они используются в качестве цитотоксического компонента в лонкастуксимабе тезирине [46]. SN-38 и дерукстекан являются ингибиторами топоизомеразы, а также входят в состав, соответственно, сацитузумаба говитекана и трастузумаба дерукстекана [47]. Помимо традиционных ингибиторов микротрубочек и агентов, повреждающих ДНК, в разработке находятся ADC с принципиально новыми механизмами действия; они расширяют терапевтические возможности этого класса препаратов. Особый интерес представляют индукторы иммуногенной клеточной смерти (ICD), такие как производные антрациклинов. Эти соединения не только непосредственно уничтожают опухолевые клетки, но и инициируют каскад иммунных реакций: способствуют экспрессии калретикулина на клеточной поверхности, высвобождению ATP и HMGB1, что в совокупности усиливает презентацию опухолевых антигенов и активацию противоопухолевого T-клеточного иммунитета [48]. Перспективным направлением являются ингибиторы транскрипции, в частности α-аманитин и его аналоги, которые селективно связываются с RNA-полимеразой II, блокируя процесс транскрипции и индуцируя апоптоз. Этот механизм демонстрирует высокую эффективность против медленно пролиферирующих опухолевых клеток и клеток с множественной лекарственной устойчивостью, что подтверждается доклиническими данными по ADC HDP-101, нацеленному на BCMA [B-клеточный антиген созревания] при множественной миеломе [49]. Еще одним инновационным классом полезных нагрузок выступают ингибиторы белков теплового шока (HSP90). Подавление HSP90 приводит к дестабилизации и последующей деградации множественных онкобелков-клиентов, включая ключевые сигнальные молекулы, участвующие в пролиферации и выживании злокачественных клеток. Данный подход особенно перспективен для лечения гетерогенных опухолей и преодоления резистентности к таргетной терапии [50]. 2. БИСПЕЦИФИЧЕСКИЕ АНТИТЕЛА: ПОВЫШЕНИЕ СЕЛЕКТИВНОСТИ ЗА СЧЕТ МУЛЬТИВАЛЕНТНОГО ТАРГЕТИНГА Другой эффективный подход к повышению селективности противоопухолевых агентов — использование биспецифических антител (bsAb). Биспецифические моноклональные антитела представляют собой генетически сконструированные белки, которые могут одновременно взаимодействовать с двумя различными мишенями. Они обладают рядом преимуществ по сравнению с обычными моноклональными антителами, среди которых: 1) перенаправление специфических поликлональных иммунных клеткок, таких как Т-клетки и NK-клетки, на опухолевые клетки для более эффективного уничтожения опухоли; 2) одновременное блокирование двух разных сигнальных путей с уникальными или перекрывающимися функциями в патогенезе; 3) увеличение специфичности связывания за счет взаимодействия с двумя различными антигенами на клеточной поверхности вместо одного. Использование препаратов bsAb для одновременного связывания и блокирования двух сигнальных путей может снизить ускользание опухолевых клеток от действия иммунной системы и улучшить терапевтическую эффективность доставляемых лекарственных средств [50]. Ключевым преимуществом bsAb для ADC является возможность повысить селективность за счет одновременного связывания с двумя различными опухолевыми антигенами. Такой подход позволяет нацеливать конъюгат на опухолевые клетки, которые ко-экспрессируют оба антигена, минимизируя воздействие на здоровые клетки, экспрессирующие лишь один из них [50]. Кроме того, биспецифические ADC могут быть сконструированы для направленной доставки в опухолевое микроокружение или для усиленной интернализации [50]. 2.1. Механизмы действия и классификация биспецифических антител Биспецифические антитела можно классифицировать по механизму их действия (рис. 3). Было создано множество bsAb для связывания Т-клеток и опухолевых клеток, которые одновременно связываются с опухолеассоциированным антигеном (ТАА), экспрессируемым на опухолевых клетках, и CD3 на Т-клетках [51]. Это перенаправляет цитотоксическую активность эффекторных Т-клеток на разрушение опухолевых клеток.
2.2. Основные мишени и терапевтические применения в онкологии К мишеням, на которые нацелены одобрнные для использования bsAb, работающие по принципу клеточных мостиков, относятся CD19, EpCAM, CD20, gp100 и BCMA. Среди bsAb, работающих по принципу клеточных мостиков и проходящих клинические испытания, мишенями являются CD33, CD37, CD38, CD123, FLT-3 и CLEC12A для гематологических злокачественных новообразований, а также HER2, CEA, PMEL, PSMA, GPA33, GPC3 и B7H3 для солидных опухолей [52]. NK-клетки также могут выступать в качестве эффекторных клеток, рекрутируемых bsAb, работающими по принципу клеточных мостиков. CD16 на NK-клетках часто используется в качестве мишени для этой категории bsAb. Например, bsAb, нацеленные на CD30×CD16, в настоящее время оцениваются в клинических испытаниях для терапии гематологических злокачественных новообразований [53]. Активно изучаются bsAb, которые могут блокировать два взаимно связанных сигнальных пути посредством нацеливания на два эпитопа на опухолевые клетки или на опухолевое микроокружение. Пары антигенов включают VEGF×Ang-2, EGFR×c-MET, IGF-1×IGF-2, VEGF×DLL4, HER2×HER2 и HER2×HER3. В мае 2021 года FDA одобрило амивантамаб (Рибревант) компании Janssen, нацеленный на EGFR×c-MET, для лечения немелкоклеточного рака легких (НМРЛ) [54]. BsAbs, которые блокируют иммунные контрольные точки, такие как PD-1×LAG3, PD-L1×TIGIT и PD-L1×CTLA-4, представляют собой еще одну категорию препаратов, протестированных в клинических испытаниях. Иммунные контрольные точки оказывают ингибирующее действие на активность иммунных клеток. bsAb, которые одновременно воздействуют на две иммунные контрольные точки, пробуют использовать для лечения солидных опухолей, поскольку они потенциально повышают эффективность ингибирования контрольных точек по сравнению с монотерапией. Кадонилимаб Akeso, блокатор PD-1×CTLA-4, был одобрен в Китае в июне 2022 года для лечения рецидивирующего или метастатического рака шейки матки. Это первый одобренный двойной ингибитор иммунных контрольных точек bsAb. Также были разработаны bsAb иммунных контрольных точек в сочетании с ТАА, такие как PD1×VEGF и PD1×HER2 [55]. BsAbs, которые совместно нацелены на костимулирующие молекулы Т-клеток и иммунные контрольные точки, такие как CTLA-4×OX40 (CD134) и PD-L1×4-1BB (CD137), в настоящее время находятся в стадии разработки [56]. Опыт создания множества успешных bsAb послужил важной основой для разработки новых. Правильный структурный дизайн и выбор мишени имеют решающее значение для обеспечения успеха разработки лекарственных средств, которые сохраняют эффективность комбинированной терапии и пониженную токсичность, что является одним из основных преимуществ биспецифических антител. Систематическое изучение успешных примеров создания биспецифических антител может помочь в разработке новых терапевтических антител. Разработка биспецифических антител открыла новые возможности в создании инновационных лекарств, которые могут воздействовать на несколько метаболических путей с уникальными или перекрывающимися функциями в патогенезе одновременно. Высокая селективность этих антител делает их идеальными для использования в качестве препаратов ADC, которые могут повысить эффективность действия против раковых клеток и увеличить интернализацию для лучшего удаления опухолевых клеток. Кроме того, ожидается, что разработка триспецифичных и тетраспецифичных антител еще больше усилит противоопухолевый эффект этих препаратов. В настоящее время проводятся клинические испытания для оценки эффективности этих препаратов [57]. Современные исследования в области биспецифических антител демонстрируют переход к созданию более сложных и селективных терапевтических платформ. Одним из наиболее значимых направлений являются триспецифические антитела, которые способны одновременно связывать два различных опухолевых антигена и иммунную клетку через рецепторы CD3 или CD16. Такие конструкции создают более точный механизм для активации иммунного ответа, существенно повышая специфичность и снижая риск неспецифических эффектов, что особенно перспективно для лечения гетерогенных опухолей [57]. Параллельно развивается направление bsAb для таргетинга онкогенных мутантных белков. Эти конструкции способны специфически распознавать опухолеспецифические мутации в таких критических онкогенах, как p53 или RAS, которые ранее считались «недосягаемыми» для терапии моноклональными антителами. Преодоление этой фундаментальной проблемы открывает новые возможности для лечения злокачественных новообразований, в развитии которых ключевую роль играют труднодоступные для лекарственного воздействия мишени, и может существенно расширить арсенал таргетной терапии [58]. Разработка этих передовых платформ bsAb представляет собой значительный шаг в эволюции иммуноонкологии, позволяя создавать высокоселективные терапевтические средства с улучшенным профилем безопасности и эффективности против наиболее сложных мишеней. На сегодняшний день все большее количество препаратов на основе биспецифических антител появляется на рынке (табл. 3).
3. МАЛЫЕ КАРКАСНЫЕ БЕЛКИ КАК АЛЬТЕРНАТИВНАЯ ПЛАТФОРМА ДЛЯ ТАРГЕТНОЙ ТЕРАПИИ Малые каркасные белки (3–20 кДа) представляют собой перспективную альтернативу, сочетающую высокое сродство к мишеням с улучшенной способностью проникать в опухолевую ткань. В отличие от антител, эти конструкции могут быть получены в бактериальных системах экспрессии, легко модифицированы и адаптированы для сайт-специфической конъюгации, что делает их идеальной основой для создания высокогомогенных терапевтических конъюгатов нового поколения. В данном разделе рассмотрены ключевые преимущества, структурное разнообразие и наиболее успешные примеры применения каркасных белков в онкологии [69].
3.1. Преимущества и ограничения каркасных белков по сравнению с традиционными антителами Большая молекулярная масса моноклональных антител снижает их проникновение в ткани солидных опухолей и препятствует связыванию внутренних антигенных эпитопов и интернализации конъюгированных с антителами лекарств [70]. Для облегчения продукции антител с полной функциональностью необходимы их посттрансляционные модификации; которые требуют использования дорогостоящей и трудоемкой эукариотической системы экспрессии, что ограничивает крупномасштабное производство и доступ к этим препаратам для большинства пациентов [71]. Более того, модификация антитела для присоединения химических линкеров и полезной нагрузки сопряжена с техническими сложностями. Кроме того, в некоторых ситуациях Fc-фрагмент может вызывать цитотоксичность, что приводит к повреждению нормальных тканей и органов. Несмотря на усилия по устранению недостатков полноразмерных антител, некоторые проблемы остались, например, более низкая авидность после модификации и необходимость гуманизации для снижения иммуногенности. Все это побудило разработчиков искать альтернативы антителам. Благодаря открытию низкомолекулярных белков с функцией связывания и постепенному развитию биотехнологий стало возможным путем мутации вариабельной области придать белкам небольшого размера способность нацеленного взаимодействия с различными белковыми мишенями [72]. Первая часть представляет собой константную область, которая включает пару α-спиралей или β-листов, составляющих жесткую третичную структуру, а второй представляет собой вариабельную область, которая образована несколькими открытыми петлями или несколькими остатками в жестких вторичных структурах. Константные области поддерживают конформационную стабильность белковых каркасов, а вариабельные области обеспечивают способность специфического связывания с различными молекулами-мишенями посредством структурного взаимодействия лиганд-рецептор или химических связей. Обычно создают библиотеки вариантов, варьируя последовательность 10-20 аминокислотных остатков в выступающих петлях, для отбора новых аффинных молекул. В белковых каркасах часто отсутствуют дисульфидные связи, и они не требуют трансляционных модификаций, что позволяет производить их с использованием экономически эффективных прокариотических систем экспрессии. Кроме того, посредством химического синтеза в белковые каркасы могут быть введены неприродные концевые аминокислотные остатки для конъюгации лекарств или диагностических реагентов. Небольшие белковые каркасы обычно растворимы в воде и обладают хорошей физико-химической стабильностью, что способствует их применению in vivo [73, 74]. Улучшенное проникновение в ткань малых каркасных белков открывает возможности для нацеливания на интрацеллюлярные мишени. Методами белковой инженерии создаются каркасы, способные проникать внутрь клетки и нарушать работу ключевых сигнальных путей, например, взаимодействующие с онкогенными белками Ras или Myc [75]. Кроме того, их малый размер позволяет использовать более экономичные методы производства, включая химический пептидный синтез, что обеспечивает легкое включение неканонических аминокислот и сайт-специфическую конъюгацию. Несмотря на множество преимуществ белковых каркасов, они больше подходят для краткосрочной визуализации рака, а не для терапии. Одна из задач для превращения таких кандидатов в реальные терапевтические агенты – усиление их фармакокинетических свойств. Представленные в обзоре малые каркасные белки характеризуются строгим соответствием системе критериев, отражающих современные требования к терапевтическим платформам. Они демонстрируют выраженное структурное разнообразие, охватывая α-спиральные, β-слоистые и смешанные архитектуры, что подтверждает широту доступных биоинженерных решений [76]. Отобранные платформы отличаются высокой технологической зрелостью, включая отработанные протоколы направленной эволюции, масштабируемого производства в бактериальных системах и сайт-специфической модификации, что критически важно для клинической трансляции [77]. Характерной особенностью представленных каркасов является доказанный терапевтический потенциал в онкологии, подтвержденный результатами доклинических исследований и данными клинических испытаний, как в случае аффибоди для визуализации HER2-позитивных опухолей и DARPins для иммуноонкологических применений [78, 79]. Особую ценность представляют уникальные функциональные преимущества данных платформ, включая превосходную степень проникновения в опухолевую ткань, возможность нацеливания на стерически затрудненные эпитопы, низкую иммуногенность и способность к формированию мультиспецифических конструкций, что целенаправленно решает ключевые ограничения терапевтических антител. Совокупность этих характеристик делает рассмотренные каркасные белки перспективной основой для создания высокоселективных противоопухолевых конъюгатов нового поколения. 3.2. Аффибоди Домен B стафилококкового белка A является родительским каркасом аффибоди, первоначально ответственного за связывание Fc иммуноглобулина G. B-домен содержит 58 аминокислотных остатков (6.5 кДа), составляющих пучок из трех α-спиралей и не содержит дисульфидных связей (рис. 4А). Затем B-домен был искусственно синтезирован и обозначен как Z-домен или аффибоди, впоследствии был использован для создания комбинаторной библиотеки путем варьирования 13 поверхностно-экспонированных остатков в первых двух спиралях [80]. Теоретически специфическое аффибоди против любой заданной мишени может быть выбрано с помощью фаговой библиотеки. Поскольку третья спираль способствует лишь структурной целостности аффибоди, но не его связывающей функции, было разработано двухспиральное аффибоди. Компенсация стабильности в этом случае достигается с помощью дисульфидных мостиков, что дает преимущества, включая меньший размер, более быстрый клиренс и меньший фоновый сигнал при визуализации[81]. Гомодимерное аффибоди ZAβ3 также использовали против амилоидных пептидов для скрининга ингибитора агрегации тау-белка. Шведская компания «Affibody AB», основанная в 1998 году, разрабатывает аффитела и владеет почти 300 патентами. Аффибоди, модифицированные контрастными веществами для визуализации, флуоресцентными метками, химическими препаратами и радиометками, применяются в терапевтических и диагностических исследованиях рака [82]. Успешное применение аффибоди продемонстрировано в препарате ABY-035 (изокибеп), где слияние анти-IL-17A аффибоди с ABD позволило добиться периода полувыведения, сопоставимого с моноклональными антителами. Отечественные исследования под руководством Долгих Д.А. и Петровская Л.Е. внесли значительный вклад в разработку стратегий применения ABD, показав, что слияние ABD с таргетными белками (например, с вариантами фибронектина, обладающими сродством к интегрину αvβ3) позволяет в несколько десятков раз увеличить период полувыведения и улучшить накопление в опухоли [83]. 3.3. Аднектин/монободи Фибронектины представляют собой макрогликопротеины, которые широко распространены у животных, отвечают за межклеточную связь и образуют внеклеточный матрикс. В качестве каркасного белка интерес представляет десятый домен фибронектина типа 3 (10FN3), имеющий структуру, подобную присутствующей в белках суперсемейства иммуноглобулинов: семь β-структур, образующих каркас в виде сэндвича, соединенных шестью петлями, три из которых являются вариабельными (рис. 4Б). Поскольку структурные особенности 10FN3 напоминают структурные особенности вариабельных тяжелых цепей антител, этот домен был выбран как основа для нового связывающего белка, названного монободи (10 кДа) [76]. Первый разработанный каркас 10FN3 против убиквитина был создан путем мутации остатков в петлях BC и FG, в то время как петля DE была введена позже для скрининга лиганда, связывающего TNF-α. Библиотека Adnectin была разработана путем рандомизации остатков в петлях BC, DE и FG 10FN3. Позже были созданы библиотеки на основе боковых петель путем изменения формы вторых β-листов CD-FG, которые образовывали вогнутый связывающий карман, подходящий для связывания с выступающими участками на поверхности глобулярных белков, таких как рецепторы [84]. Наиболее продвинутым клиническим кандидатом является PRS-343 (сигалинцин-аднектин) – биспецифическая молекула, направленная на HER2 и рецептор 4-1BB (CD137) для активации противоопухолевого иммунитета в микроокружении HER2-позитивных опухолей [85]. Препарат демонстрировал обнадеживающие результаты в клинических испытаниях I фазы (NCT03330561) [86]. 3.4. Смоделированные белки с анкириновыми повторами (DARPins) Мотив анкиринового повтора (AR) с конформацией спираль-петля-спираль входит в состав многих бактериальных, архейных и эукариотических белков, но гораздо чаще встречаются у эукариот и участвуют исключительно в опосредовании белок-белковых взаимодействий, включая транскрипцию, транспорт ионов и модуляцию клеточного цикла (рис. 4В). Анализ кристаллической структуры и консенсусной последовательности анкиринового мотива показал, что консервативное гидрофобное ядро, содержащее α-спирали, представляет собой жесткую структуру, в то время как менее консервативная петлевая область способствует молекулярному распознаванию, что лежит в основе конструкции белкового центра связывания [87]. Каркасы DARPin включают 2–3 мотива анкиринового повтора человека, фланкированные кэпирующими повторами как на N-, так и на C-концах, которые защищают их от агрегации во время процесса сворачивания. Каждая единица анкиринового повтора (33 аминокислоты) образует структурный мотив из двух антипараллельных α-спиралей, соединённых петлёй и β-поворотом; концевые участки этого мотива обеспечивают взаимодействие с соседними доменами [88]. В соответствии с консенсусной стратегией разработки, биофармацевтическая компания «Molecular Partners AG», основанная в 2004 году, разработала новые молекулы DARPin в качестве альтернативы антителам. Кроме того, была создана библиотека петли DARPin с введенным доменом антитела CDR-H3 для расширения связывающей способности молекул [89]. В литературе сообщалось о более чем 60 молекулах DARPin. Благодаря их небольшому размеру (14–18 кДа), физико-химической стабильности и простоте получения поливалентных или мультиспецифических элементов таргетинга, DARPin стали привлекательны для диагностических и терапевтических применений [90]. Отечественные исследования под руководством Деева С.М. вносят значительный вклад в развитие тераностической платформы на основе DARPins, нацеленной на рецептор HER2 (человеческий эпидермальный фактор роста 2). Продемонстрирована возможность создания высокоаффинного конъюгата DARPin 9_29 с золотыми наностержнями (GNRs). Такой конъюгат специфично связывается с клетками аденокарциномы молочной железы SK-BR-3, сверхэкспрессирующими HER2, и приводит к их практически полной эрадикации при облучении ближним инфракрасным светом, обладая при этом селективным действием. В другом исследовании тот же DARPin 9_29 был успешно использован в качестве основы для радионуклидного зонда, меченного йодом-125 или технецием-99m, для молекулярной визуализации экспрессии HER2. Зонд продемонстрировал высокую специфичность, аффинность и значимое накопление в HER2-положительных ксенографтах, причем радиоизотоп йода показал более благоприятный профиль биораспределения. Эти работы подтверждают перспективность модульной архитектуры DARPins для создания высокоспецифичных конъюгатов как для таргетной терапии, так и для диагностической визуализации [91, 92]. 3.5. Аффимеры Другой каркас, аффимеры (~14 кДа), происходит от ингибиторов протеазы стефина А человека, включающие в себя α-спираль, а также четыре антипараллельных β-листа и две неструктурированные петли (рис. 4Д) [93]. Платформа аффимеров Affimer® разрабатывается компанией Avacta Group plc для создания новых терапевтических и диагностических средств. В терапевтической области фокус сделан на разработке аффимерно-цитокиновых конъюгатов и биспецифичных аффимеров для таргетной иммунотерапии онкологических заболеваний. В диагностике технология применяется для создания высокоспецифичных реагентов, которые могут использоваться для визуализации и анализа in vitro [94]. 3.6. Альбумин-связывающий домен (ABD) как каркасный белок Альбумин-связывающий домен (ABD) представляет собой малый (около 5 кДа) и высокостабильный белковый каркас, производный от белков клеточной стенки бактерий рода Streptococcus (рис. 4Г). Его ключевой особенностью является сродство с Кд в пикомолярном диапазоне концентраций к сывороточному альбумину человека (HSA) [95]. Эта уникальная особенность не используется для прямого нацеливания на опухоль, а служит для существенного улучшения фармакокинетических параметров терапевтических молекул. При слиянии ABD с целевыми белками (например, сцинтилляционными маркерами, цитокинами или самими каркасными белками) образуется долгоживущий комплекс с эндогенным альбумином, что защищает конструкцию от быстрого почечного клиренса и продлевает ее период полувыведения из крови, имитируя длительный период полураспада антител, которые содержат Fc-фрагмент [96]. В контексте онкологии домен, связывающий альбумин (ABD), активно используется для увеличения периода полувыведения и улучшения фармакокинетики целевых терапевтических молекул, таких как аффибоди и другие каркасные белки. Классическим примером успешного применения технологии ABD за пределами онкологии является идаруцизумаб (Прадакса®) – специфический антидот, в структуру которого интегрирован ABD для пролонгации времени циркуляции и эффективного связывания антикоагулянта. Этот же принцип переносится в область противоопухолевой терапии [97]. Технология основана на способности ABD обратимо связываться с сывороточным альбумином, что значительно замедляет клиренс конъюгата и способствует его пассивному накоплению в опухолевой ткани. В онкологии ABD конъюгируют с цитотоксическими агентами (радиоизотопами, цитотоксинами) для создания препаратов с улучшенной опухолевой экспозицией и терапевтическим индексом. Примером служат аффибоди, нацеленные на HER2 и конъюгированные с ABD, которые разрабатываются для радионуклидной терапии [98]. На основе альбумин-связывающего домена (ABD) был создан новый радиофармпрепарат ABY-028, меченный галлием-68, и с помощью позитронно-эмиссионной томографии (ПЭТ) изучено его накопление в зависимости от проницаемости эндотелия [99]. 4. СТРАТЕГИИ САЙТ-СПЕЦИФИЧЕСКОЙ КОНЪЮГАЦИИ В РАЗРАБОТКЕ АНТИТЕЛ-ЛЕКАРСТВЕННЫХ И ПЕПТИД-ЛЕКАРСТВЕННЫХ КОНЪЮГАТОВ ДЛЯ ОНКОЛОГИИ Стратегии создания конъюгатов антител с цитотоксическими препаратами (ADC) в целом подразделяются на два фундаментальных типа: традиционную (несайт-специфическую) и сайт-специфическую конъюгацию. В основе обоих подходов лежит формирование ковалентных связей между моноклональным антителом и цитотоксическим агентом. Этот процесс, как правило, опосредован реакционноспособными аминокислотными остатками, экспонированными на поверхности иммуноглобулина, преимущественно лизином и цистеином [100]. 4.1. Конъюгация по остаткам лизина Конъюгация по остаткам лизина представляет собой широко распространенный метод синтеза ADC, основанный на реакционной способности нуклеофильных ε-аминогрупп (--NH2) доступных остатков лизина (рис.5Б). Данные аминогруппы обладают высокой реакционной способностью в условиях, близких к нейтральным, что обеспечивает их селективное взаимодействие с линкерными молекулами, несущими электрофильные группы [101]. Такой подход обеспечивает эффективное ковалентное присоединение полезной нагрузки к антителу, однако часто приводит к образованию гетерогенных продуктов вследствие высокой распространенности и неравномерного распределения остатков лизина на поверхности иммуноглобулина [102]. Электрофильные реагенты преимущественно атакуют нуклеофильные ε-аминогруппы лизина, что позволяет осуществлять конъюгацию без необходимости предварительной модификации каркаса антитела [101]. Все это стимулировало разработку разнообразных гетеробифункциональных электрофильных линкеров, позволяющих контролировать процесс конъюгации. В отличие от классических гомобифункциональных реагентов (например, глутарового альдегида), склонных к неспецифическому сшиванию и образованию гетерогенных продуктов, для создания терапевтических ADC применяются специализированные электрофильные соединения. К числу широко используемых относятся сложные эфиры N-гидроксисукцинимида (NHS) и их производные, изотиоцианаты и сквараты [103]. Однако, несмотря на высокую реакционную способность, данные линкеры (в особенности NHS-эфиры) подвержены гидролизу в водных средах, что может снижать эффективность конъюгации и выход целевого продукта [104].
Современное развитие метода направлено на преодоление этих ограничений через переход от стохастической к управляемой конъюгации. Исследования выявили, что при строгой стандартизации производственных условий распределение полезной нагрузки по остаткам лизина оказывается не случайным, а воспроизводимым, что открывает путь к контролируемому и масштабируемому производству. Дальнейшая эволюция заключается в разработке стратегий сайт-специфической конъюгации с остатками лизина. Использование линкеров с умеренной электрофильностью (таких как β-лактамы, метилсульфоны) или специальных платформ (например, K-lock™) позволяет направленно модифицировать наиболее реакционноспособные или структурно предопределенные остатки лизина (например, Lys99, Lys188). Эти усовершенствования ведут к получению более гомогенных популяций ADC с предсказуемым DAR, улучшенной стабильностью и оптимизированными фармакологическими свойствами.
4.2. Конъюгация по сульфгидрильным группам цистеина с использованием малеимидов Одним из наиболее распространенных методов, наряду с конъюгацией по остаткам лизина, является стохастическая конъюгация по нативным остаткам цистеина. Этот подход основан на частичном восстановлении четырех межцепочечных дисульфидных связей в молекуле IgG1, что приводит к экспозиции реакционноспособных тиоловых групп (--SH). Эти тиолы вступают в реакцию Михаэля с электрофильными акцепторами, чаще всего с малеимидными производными синтетических цитотоксических агентов (рис. 5А). Варьируя условия восстановления и стехиометрию полезной нагрузки, можно получать ADC с различным и контролируемым коэффициентом конъюгации (DAR), преимущественно равным 2, 4, 6 или 8 [107]. Однако, несмотря на управляемость DAR, этот метод, как и конъюгация по остаткам лизина, порождает гетерогенную популяцию молекул, являющихся региоизомерами из-за присоединения к разным остаткам цистеина. Эта гетерогенность негативно сказывается на терапевтическом индексе, поскольку разные изомеры обладают вариабельными фармакокинетическими профилями, токсичностью и склонностью к агрегации. Более контролируемым вариантом данной технологии является использование инженерных цистеинов. Метод заключается во внесении точечных мутаций для кодирования остатков цистеина в строго заданных позициях антитела, что позволяет направленно присоединять полезную нагрузку. Это обеспечивает не только однородный DAR, но и контроль над ориентацией цитотоксического агента, что было реализовано, например, в препарате лонакатуксимаб тезиринейн (DAR2) [108]. Подобный подход способствует получению более гомогенного и воспроизводимого продукта. Одним из существенных недостатков классической тиол-малеимидной химии является обратимость реакции Михаэля (ретро-реакция Михаэля), скорость которой зависит от положения конъюгации. Это может приводить к преждевременному высвобождению полезной нагрузки in vivo и её неспецифическому переносу на другие тиолсодержащие молекулы, например, на альбумин или глутатион, что усиливает внецелевую токсичность. Для повышения стабильности связи разработаны стратегии, приводящие к гидролизу сукцинимидного кольца с образованием более стабильных и необратимых аддуктов. Логическим развитием стремления к гомогенности стало создание ADC с DAR8 путем конъюгации по всем межцепочечным цистеинам. Однако, несмотря на улучшение in vitro эффективности, такие высоконагруженные конъюгаты демонстрируют сниженную in vivo активность из-за ускоренного клиренса, вызванного повышенной гидрофобностью и нарушением важных структурных дисульфидных связей [108]. Альтернативой стал метод тиолового мостика, при котором бифункциональные конструкции «линкер-полезная нагрузка» используются для сшивания восстановленных межцепочечных цистеинов. Этот подход позволяет получать гомогенные ADC с DAR4, восстанавливая при этом нативную дисульфидную сеть, что повышает стабильность антитела, улучшает фармакокинетику и переносимость in vivo по сравнению с традиционными конъюгатами. Несмотря на эти недостатки, большинство одобренных ADC по-прежнему используют стохастические методы конъюгации. Их ключевые преимущества – простота и скорость производственного процесса, так как они не требуют сложной белковой инженерии и используют нативные последовательности антител [109]. Для создания более однородных ADC был разработан метод thiomAb, который позволяет проводить сайт-специфическую конъюгацию по искусственно введенным остаткам цистеина. Процесс включает три этапа: восстановление для раскрытия тиольных групп, окисление для реформирования нативных дисульфидных мостиков и, наконец, конъюгацию к введенным остаткам цистеинам. Этот метод сохраняет структурную целостность антитела и предоставляет возможность стратегического выбора позиции для конъюгации с целью оптимизации гидрофобности и стабильности конечного продукта. Систематический скрининг различных позиций показал, что эффективность конъюгации и стабильность ADC сильно зависят от выбранного сайта, и лишь немногие позиции обеспечивают желаемые свойства. Интересно, что были идентифицированы мутантные цистеины (например, замена Gln124 на Cys в легкой цепи), которые обладают высокой реакционной способностью и могут конъюгироваться без предварительного этапа восстановления-окисления, что потенциально упрощает производство [110]. 4.3. Ферментативная конъюгация и клик-химия Ферментативная конъюгация представляет собой высокоточную технологию, использующую ферменты для ковалентного присоединения полезной нагрузки к антителам в строго определенных аминокислотных последовательностях, что позволяет получать высокогомогенные ADC с заданными сайтами конъюгации. Данная стратегия доказала свою эффективность в качестве метода конъюгирования, стабильно обеспечивая получение однородных популяций ADC. Однако многие ферментативные платформы требуют модификации последовательности или структуры антитела для введения совместимых мотивов узнавания [111]. Например, транспетидаза Сортаза A (SrtA), широко применяемая в разработке ADC, расщепляет мотив LPXTG между остатками треонина и глицина. Образовавшийся ацильный фрагмент ковалентно связывается с остатком цистеина в активном центре фермента через тиоэфирную связь, формируя ацил-ферментный интермедиат. Этот интермедиат затем вступает в реакцию транспептидации с N-концевой аминогруппой субстрата-акцептора, содержащего олигоглициновый мотив, что приводит к переносу полезной нагрузки на акцептор [111]. Клинически значимым примером фермент-опосредованной конъюгации является ADC TRPH-222, в настоящее время проходящий испытания I фазы, который использует платформу SMARtag™ на основе технологии формилглицин-генерирующего фермента (FGE) для введения альдегидных меток с целью сайт-специфического присоединения нагрузки [111]. Технология FGE специфически распознает мотивы CXPXR. Она катализирует окисление остатка цистеина в этой последовательности до формилглицина, вводя уникальную альдегидную функциональную группу. Эта альдегидная метка затем селективно реагирует с линкерами, содержащими, например, аминоокси- или гидразиновые группы, обеспечивая сайт-специфическую биоконъюгацию. Другой фермент-зависимый подход использует белок-фарнезилтрансферазу (PFTase), которая присоединяет изопреноидные липидные группы (например, фарнезил) к остаткам цистеина в мотиве CaaX-бокса. Модифицированные изопреноидные группы в свою очередь служат субстратами для последующих биоортогональных реакций (например, клик-химии), позволяя проводить хемоселективные конъюгации. Клинический пример, использующий эту технологию, – FS-1502, разработанный с использованием платформы ConjuALL™ и проходящий в настоящее время клинические испытания III фазы [112]. В совокупности биоконъюгация, опосредованная FGE и PFTase, представляет собой мощные фермент-управляемые методы для создания ADC нового поколения с улучшенной сайт-специфичностью, воспроизводимостью между партиями и терапевтическим индексом. В настоящее время в доклинических исследованиях для разработки ADC изучаются несколько ферментных систем конъюгации. К ним относятся пептидаспарагиниллигазы, тубулин-тирозиллигазы, трипсилигазы, фосфопантетеинилтрансферазы, SpyLigase и O6-алкилгуанин-ДНК-алкилтрансфераза, широко известная как SNAP-тег [113]. Эти ферменты предлагают разнообразные механизмы сайт-специфического присоединения нагрузки за счет распознавания уникальных пептидных последовательностей или химических мотивов [114]. Среди наиболее продвинутых ферментативных платформ микробная трансглутаминаза (mTG) катализирует образование изопептидных связей между γ-карбоксамидной группой остатков глутамина и первичными аминами [113]. В контексте ADC mTG селективно нацеливается на определенные нативные сайты на антителах, таких как остаток Q295 в Fc-области. Ключевым клиническим примером является DP303, в настоящее время проходящий испытания III фазы, который использует mTG для эффективной конъюгации, позволяя свободной аминогруппе полезной нагрузки реагировать с сайтом Q295, что приводит к получению высокостабильных и гомогенных ADC с благоприятными фармакокинетическими и терапевтическими свойствами [113]. Успешная разработка фермент-конъюгированных ADC требует тщательной оптимизации как фармакокинетических свойств конечного продукта, так и масштабируемости и воспроизводимости процесса конъюгации. Клик-химия предлагает многочисленные преимущества для синтеза ADC, включая мягкие условия реакции, высокие выходы, совместимость с водными и органическими растворителями, стереоселективность и простоту выделения продукта, что делает её высоко подходящей для задач биоконъюгации [116]. В результате клик-реакции стали мощным и эффективным инструментом для биоконъюгации – процесса ковалентного связывания химически различных молекул – благодаря своей универсальности, надежности и совместимости со сложными биологическими системами [117]. Не все клик-реакции изначально подходят для биологических систем; для оптимальных результатов в биоконъюгации реакция должна быть биоортогональной, то есть проявлять минимальную или нулевую реакционную способность с эндогенными функциональными группами, чтобы сохранять нативную биологическую среду [118]. В 2002 году Торнё, Кристенсен и Мельдал впервые применили принципы клик-химии для биоконъюгации двух биологических молекул, используя твердофазный синтез для получения пептидотриазолов посредством региоспецифичной медь(I)-катализируемого азид-алкиновойго циклоприсоединения (CuAAC) терминальных алкинов с азидами, что ознаменовало собой ключевой прорыв в области сайт-селективного биомолекулярного связывания [119]. Биоконъюгация стала критически важным инструментом в различных научных областях, включая диагностику, терапию и материаловедение. В контексте разработки ADC клик-химия может обеспечить эффективное и селективное связывание биологически активных компонентов [120]. Ключевые клик-стратегии, используемые в синтезе ADC, многочисленны и включают тиол-малеимидное присоединение по Михаэлю, медь-катализируемое азид-алкиновое (3+2) циклоприсоединение (CuAAC) (рис. 6А), образование оксимной связи (рис. 6Б), классическую реакцию Дильса–Альдера (рис. 6В), гидразино-изо-лигирование по Пикте–Шпенглеру (HIPS) и реакцию Дильса-Альдера с обратным переносом электрона, каждая из которых предлагает уникальные преимущества с точки зрения специфичности реакции, биоортогональности и стабильности конъюгата [121].
В разработке ADC такие клик-реакции, как CuAAC, образование оксимов и реакция Дильса–Альдера, предлагают универсальные, биоортогональные и эффективные стратегии для стабильной и сайт-специфической биоконъюгации. 5. ПЕРСПЕКТИВНЫЕ ПЛАТФОРМЫ ДЛЯ ТАРГЕТНОЙ ТЕРАПИИ: ОТ ПРОТЕОЛИТИЧЕСКОЙ ДЕГРАДАЦИИ ДО УПРАВЛЯЕМЫХ КОНЪЮГАТОВ Парадигма разработки таргетных противоопухолевых препаратов вышла за рамки простой доставки цитотоксических агентов. Современные исследования сосредоточены на создании принципиально новых платформ, которые действуют на ранее «недосягаемые» мишени и обеспечивают беспрецедентный уровень контроля над терапевтическим воздействием. Эти подходы, многие из которых находятся на переднем крае науки, формируют основу для терапии следующего поколения. 5.1. PROTAC-ADC (Degrader-Antibody Conjugates) Развитие терапий, направленных на деградацию болезнетворных белков, представляет собой одну из самых динамичных областей современной разработки препаратов. Традиционная парадигма, в которой малые молекулы-ингибиторы временно блокируют активность мишени, имеет фундаментальные ограничения. Она плохо применима к обширному классу белков, которые традиционно считаются труднодоступными для таргетной терапии из-за отсутствия чётких активных сайтов, таких как транскрипционные факторы или каркасные белки [122]. Ответом на этот вызов стала технология PROteolysis TArgeting Chimeras (PROTAC), использующая принципиально иную, event-driven фармакологию. PROTAC представляют собой гетеробифункциональные молекулы, состоящие из лиганда к белку-мишени (POI), лиганда к E3 убиквитин-лигазе и соединяющего их линкера [123]. Их каталитический механизм действия заключается в формировании тернарного комплекса POI–PROTAC–E3, что приводит к полиубиквитинизации POI и его последующей деградации 26S протеасомой [124] Этот подход доказал свои ключевые преимущества: возможность воздействовать на белки, которые считаются «нелегируемыми» для традиционных ингибиторов, благодаря каталитическому механизму деградации [125], а также способность преодолевать резистентность, вызванную мутациями мишени (например, BTK C481S [126] или AR [127]), и возможность полностью устранять все функции белка, включая каркасные, что критично для таких мишеней, как рецепторные тирозинкиназы [128]. Однако широкому терапевтическому применению классических PROTAC препятствуют серьезные ограничения. Главным из них является отсутствие тканевой и клеточной селективности, поскольку используемые E3 лигазы (VHL, CRBN и др.) экспрессируются повсеместно [129]. Кроме того, PROTAC часто обладают неоптимальными фармакокинетическими свойствами: высокая молекулярная масса (700–1100 Да) и полярность ведут к низкой пероральной биодоступности и быстрому клиренсу, требуя сложных формуляров для исследований in vivo. Для решения, в первую очередь, проблемы селективности была разработана стратегия конъюгации PROTAC с моноклональными антителами, что привело к созданию нового класса соединений – Degrader-Antibody Conjugates (DAC), также известных как Antibody-PROTAC Conjugates [130]. Эта платформа объединяет преимущества двух технологий: точную таргетную доставку антитела к специфическому поверхностному антигену (например, HER2, CLL1, CD22) и каталитическую эффективность PROTAC. Конъюгат антитело-PROTAC действует по принципу таргетной доставки гетеробифункциональной молекулы, направляющей белок-мишень на деградацию, внутрь специфических клеток. Процесс начинается со связывания антительной части конъюгата с поверхностным антигеном целевой клетки. После связывания комплекс интернализуется путём эндоцитоза, последовательно проходя стадии ранней и поздней эндосомы, а затем попадая в лизосому. В лизосомальных условиях специальный линкер расщепляется, высвобождая активную молекулу PROTAC в цитоплазму. Здесь PROTAC выполняет свою основную функцию: одна его часть связывает белок-мишень (POI), а другая — E3 убиквитин-лигазу, формируя тройной комплекс. Это приводит к полиубиквитинированию белка-мишени, который затем распознаётся и необратимо разрушается 26S протеасомой (рис.7). Таким образом, данная платформа объединяет точность антительной доставки с мощным каталитическим механизмом направленной деградации внутриклеточных белков, что особенно перспективно для воздействия на «недосягаемые» мишени в онкологии. [131]), что гарантирует ее проявление только после внутриклеточного высвобождения. Практическая реализация концепции DAC была успешно продемонстрирована на нескольких моделях. Например, конъюгат CLL1-DAC, доставляющий соединение, направленное на деградацию BRD4 GNE-987 в клетки острого миелоидного лейкоза, преодолел неблагоприятную фармакокинетику свободного соединения и показал эффективность in vivo [132]. Другой пример – HER2-DAC, нацеленный на деградацию эстрогенового рецептора (ERα) в HER2-позитивных раковых клетках, где в качестве линкера использовался ферментативно расщепляемый пептид (Val-Cit-PABC) [133]. Эти примеры иллюстрируют важность оптимизации таких параметров, как стабильность линкера в кровотоке и Drug-to-Antibody Ratio (DAR), высокое значение которого (>4) часто необходимо для эффективности, но может осложнять свойства конъюгата [134]. Перспективы технологии DAC связаны с потенциалом создания высокоселективных терапевтических подходов с широким терапевтическим окном. Однако ее развитие сталкивается с рядом вызовов. К ним относятся необходимость расширения арсенала лигандов E3, особенно с тканеспецифичной экспрессией [135], глубокое понимание биологии для выбора согласованных пар «поверхностный антиген – внутриклеточная мишень», а также комплексная оптимизация фармакокинетики и профиля безопасности, включая контроль потенциальной нецелевой деградации [127]. Клинический прогресс, достигнутый в разработке индукторов направленной деградации белка, подтверждает общую жизнеспособность этого подхода и открывает путь для будущих клинических испытаний самих DAC. [136].
Таким образом, эволюция от PROTAC к DAC представляет собой логический и необходимый шаг в стремлении к истинно прецизионной медицине. PROTAC заложили революционную основу, переведя фармакологию из режима временного ингибирования в режим полного устранения белка. DAC, в свою очередь, решают ключевое ограничение первых, обеспечивая доставку этого мощного каталитического инструмента строго в целевые клетки. Эта конвергенция технологий открывает новую главу в разработке терапевтических средств, обладающих потенциалом для лечения заболеваний, ранее считавшихся трудно поддающимися терапии. 5.2. Олигонуклеотидные конъюгаты: подавление онкогенов на уровне РНК Антитело-олигонуклеотидные конъюгаты (AOCs) представляют собой инновационный класс таргетных биологических препаратов, развивающих успешную концепцию антитело-лекарственных конъюгатов (ADCs). Ключевое отличие заключается в терапевтической нагрузке: вместо цитотоксических молекул AOCs доставляют в опухолевые клетки регуляторные олигонуклеотиды, такие как малые интерферирующие РНК (siRNA) и антисмысловые олигонуклеотиды (ASO). Это позволяет перейти от стратегии уничтожения клеток к прецизионной регуляции экспрессии генов, лежащих в основе онкогенеза. Конъюгат формируется на модульной основе и состоит из трех основных компонентов: таргетного моноклонального антитела (или его фрагмента), обеспечивающего специфическое связывание с антигеном на поверхности раковой клетки и последующую интернализацию; химического линкера (расщепляемого или нерасщепляемого), который ковалентно соединяет носитель с нагрузкой; и собственно терапевтического олигонуклеотида, последовательность которого комплементарна мРНК целевого онкогена [136]. Механизм действия AOCs направлен на подавление экспрессии онкогенов на уровне РНК и реализуется по двум основным путям в зависимости от типа нагрузки. Конъюгаты на основе siRNA используют клеточный аппарат RNA-интерференции. После интернализации и высвобождения двуцепочечная siRNA загружается в РНК-индуцированный silencing-комплекс (RISC), который направляется к комплементарной последовательности матричной РНК онкогена, вызывая ее расщепление и деградацию, что блокирует синтез онкобелка [137]. ASO-содержащие конъюгаты, будучи одноцепочечными, гибридизуются с целевой мРНК, что приводит либо к рекрутированию нуклеазы RNase H1 и разрушению транскрипта, либо к стерическому блокированию сайта связывания рибосомы или процессинга, подавляя трансляцию или изменяя сплайсинг. Для повышения стабильности, аффинности и снижения иммуногенности терапевтические олигонуклеотиды подвергаются химическим модификациям, таким как введение фосфоротиоатных связей и замещение 2'-OH группы на 2'-O-метил или 2'-фтор [137]. Ключевым преимуществом платформы AOC является синергия высокой специфичности антител и программируемости олигонуклеотидов. Это позволяет преодолевать фундаментальные ограничения доставки нуклеиновых кислот во внепеченочные ткани и таргетно воздействовать на онкогены, считающиеся «недосягаемыми» для малых молекул. Платформа предлагает универсальный подход для подавления широкого спектра молекулярных мишеней, участвующих в пролиферации, выживании и метастазировании опухолевых клеток. Однако клинической трансляции AOC в онкологии препятствуют серьезные вызовы. К ним относятся необходимость эффективного выхода олигонуклеотида из эндосомы в цитоплазму для доступа к аппарату RISC, риск внецелевых эффектов из-за частичной гомологии последовательностей, сложности в производстве гомогенных конъюгатов и ограниченные данные по фармакокинетике и токсикологии in vivo [137]. В настоящее время большинство AOC на продвинутых стадиях клинических испытаний разрабатываются для лечения нейромышечных заболеваний, таких как миотоническая дистрофия 1 типа (AOC 1001) [137]. В онкологической практике одним из первых кандидатов является TAC-001 от Tallac Therapeutics – конъюгат анти-CD22 антитела с агонистом толл-подобного рецептора 9 (CpG-олигонуклеотидом), находящийся в фазе I/II исследований для лечения B-клеточных злокачественных новообразований [137]. Многочисленные доклинические исследования демонстрируют потенциал AOC против различных онкогенов (c-Myc, KRAS, STAT3, PLK1) в моделях солидных опухолей и гематологических злокачественных новообразований. Таким образом, олигонуклеотидные конъюгаты открывают новую терапевтическую парадигму для подавления онкогенов на уровне РНК, хотя их полный клинический потенциал в онкологии станет очевиден по мере преодоления существующих технологических барьеров и накопления данных клинических испытаний. ЗАКЛЮЧЕНИЕ Таким образом, вектор развития таргетной терапии направлен на создание интеллектуальных мультифункциональных тераностических агентов. Однако путь к их широкой клинической реализации сопряжён с необходимостью преодоления специфических барьеров, присущих каждой платформе. Для конъюгатов антитело-лекарство (ADC) ключевой вызов заключается в оптимизации терапевтического индекса, что требует преодоления гетерогенности препаратов с помощью сайт-специфической конъюгации и точного балансирования между способностью цитотоксической нагрузки распространяться на соседние клетки опухоли (включая клетки с низкой экспрессией антигена) и риском повреждения здоровых тканей. Биспецифические антитела, в свою очередь, нуждаются в решении проблем иммуногенности и контроле потенциально жизнеугрожающих побочных эффектов, таких как синдром высвобождения цитокинов (CRS), а также в поиске оптимальных пар антигенов для достижения истинной опухолевой селективности. Платформы на основе малых каркасных белков сталкиваются с необходимостью улучшения фармакокинетики, например, путём слияния с альбумин-связывающим доменом (ABD), и получения убедительных доказательств их клинической эффективности в терапии солидных опухолей. Конъюгаты антитело-PROTAC (DAC) стоят перед комплексными проблемами обеспечения in vivo стабильности и активности сложной гетеробифункциональной конструкции, контроля внецелевой деградации белков и достижения высокого коэффициента конъюгации (DAR) без ущерба для фармакокинетических свойств. Для олигонуклеотидных конъюгатов (AOC) принципиальным ограничением остаётся низкая эффективность выхода полезной нагрузки из эндосом в цитоплазму, а также вопросы долгосрочной стабильности и иммуногенности олигонуклеотидов. Преодоление этих многоуровневых вызовов потребует конвергенции усилий в области белковой инженерии, химии линкеров, фармакологии и дизайна клинических исследований. Интеграция методов искусственного интеллекта для in silico предсказания свойств конъюгатов и идентификации новых мишеней станет критическим фактором ускорения разработки. Успешное решение указанных задач приблизит реализацию парадигмы истинно персонализированной онкологии, в основе которой лежат агенты, способные не только к избирательному уничтожению опухолевых клеток, но и к преодолению механизмов резистентности и тонкой модуляции противоопухолевого иммунного ответа. СОБЛЮДЕНИЕ ЭТИЧЕСКИХ СТАНДАРТОВ Настоящая статья не содержит каких-либо исследований с участием людей или с использованием животных в качестве объектов исследования. ФИНАНСИРОВАНИЕ Работа выполнена в рамках государственного задания Министерства науки и высшего образования Российской Федерации по теме «Технологическая платформа для разработки лекарственных кандидатов на основе альтернативных каркасных белков для высокоселективнойдоставки цитотоксических агентов в раковые клетки» проект (No FMRM-2025-0016) КОНФЛИКТ ИНТЕРЕСОВ Авторы заявляют об отсутствии конфликта интересов ЛИТЕРАТУРА
|